Организм человека — очень сложная система, малейший сбой которой может повлечь за собой изменения в работе многих систем.
Зачастую ребенок только успел появиться на свет, а его уже сопровождают неприятные заболевания. К сожалению, природу не обманешь, и нарушения на генном уровне могут проявляться с первых дней жизни.
Так, диагноз буллезный эпидермолиз относится именно к таким заболеваниям.
Особенности диагноза буллезный эпидермолиз и его симптомы
Буллезный эпидермолиз достаточно редкое заболевание — встречается у одного из десятков или сотен тысяч пациентов. Как правило, оно проявляется с самого рождения и в зависимости от разновидностей его симптомы с возрастом могут угасать, или становиться более явными. Зачастую болезнь может затрагивать и другие жизненно-важные системы организма, а также сопровождаться развитием инфекции.
Особенность состоит в том, что мутации затрагивают гены в различных слоях кожного покрова, которые кодируют белки. При этом изменения могут происходить не в одном гене, а сразу в нескольких — зачастую болезнь нарушает структуру более 10 генов.
Отличительными признаками данного диагноза являются следующие симптомы:
- наличие на коже пузырей, наполненных жидкостью без цвета и запаха, которые заживают без рубцов, или оставляя их;
- появление эрозии кожи;
- повышенная чувствительность кожных покровов и склонность к появлению травм, трещин и микроповреждений при малейшем воздействии;
В зависимости от тяжести и типа буллезного эпидермолиза возможны и другие, более тяжелые симптомы, которые рассматриваются в индивидуальном порядке.
Симптомы зачастую проявляются в первые часы жизни новорожденного, поэтому квалификация и осведомленность медицинского персонала будет немаловажным в дальнейшем лечении пациента.
Причина возникновения эпидермолиза буллезного
Основная причина — генетическая предрасположенность к мутации генов, отвечающих за кодирование белков, которая передается по наследству от родителей к детям.
Как правило, это происходит двумя способами:
- Аутосомно-доминантный — характерен для простого типа буллезного эпидермолиза.
- Аутосомно-рецессивный — для пограничного.
Еще один тип — дистрофический — может наследоваться обоими способами.
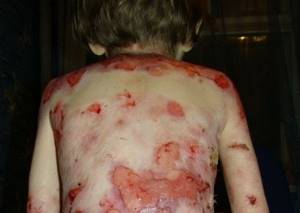
Мужчины и женщины имеют одинаковую степень риска проявления данной болезни. Это достаточно редкое явление, но его примеры существуют. Данный опыт используется для научных исследований и разработки методов лечения.
При планировании ребенка важно уточнить информацию о наличии у родителей наследственных заболеваний, возможно, пройти некоторые генетические исследования, которые позволят предупредить развитие серьезных патологий. Также родителям из группы риска при установлении факта беременности желательно проходить все запланированные обследования и скрининги для выявления отклонений в развитии плода.
Различные формы эпидермолиза
Классификация буллезного эпидермолиза основана на возможности появления пузырей на различных уровнях слоев кожи.
Каждая форма разделяется на несколько подвидов в зависимости от следующих факторов:
- генотипа;
- фенотипа;
- принципа наследования.
Выделяют несколько различных форм.
Простой вид
Ему свойственно образование пузырей в верхних слоях кожи. Их локализация — стопы, кисти рук, в тяжелых случаях может быть поражено все тело человека. При легкой степени поражения пузыри проходят без появления рубцов, но из-за чрезмерной чувствительности кожи возможно появление рецидивов.
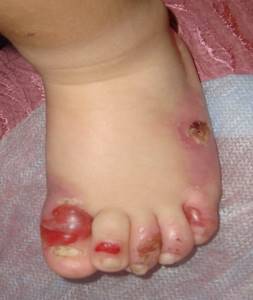
Среди подтипов простого вида выделяют два основных:
- Локализованный — при котором обострение эпидермолиза происходит в летний период. Пузыри появляются на ладонях, стопах или по всему телу, но с возрастом их количество, как правило, снижается. Болезнь может сопровождать несколько поколений.
- Генерализованный тип Доулинга-Меары — одна из самых тяжелых форм буллезного эпидермолиза. Она характеризуется появлением пузырей уже с самого рождения.
Дополнительными симптомами могут быть:
- гиперпигментация;
- некоторые разновидности гиперкератоза;
- мили (мелкие узелки белого цвета);
- нарушение слизистых оболочек;
- появление рубцов;
- дистрофия ногтевых пластин, сращение пальцев;
- заболевания сердечно-сосудистой системы;
- нарушение развития и задержка роста.
Данная разновидность зачастую имеет неблагоприятный прогноз — высока смертность новорожденных с таким диагнозом.
Дистрофический вид
Характеризуется появлением пузырей на уровне верхних частей сосочкового слоя кожи, но при этом ниже уровня плотной пластины. Локализация пузырей — в основном руки и ноги, иногда все тело.
Симптомы в большинстве случаев стандартны — эрозия, пузыри, рубцевание, дистрофия или утрата ногтевых пластин. Часты рецидивы заболевания.
В большинстве случаев отличается тяжелым поражением кожных покровов.
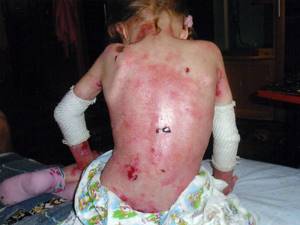
Различают два основных подтипа в зависимости от вида наследования:
В зависимости от подтипа диагноза возможны следующие осложнения:
- задержка развития;
- появление контрактуры суставов — ограничение их движения;
- проблемы с функционированием желудочно-кишечного тракта;
- анемия;
- заболевания роговицы глаз;
- высокий риск развития онкологических заболеваний.
Пограничный вид
Для этого вида характерно появление пузырей в слоях кожи, которые соответствуют уровню светлой пластины кожи.
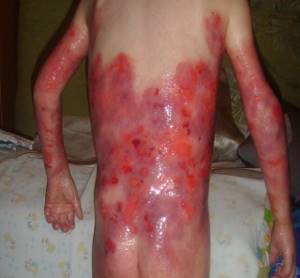
Его особенности:
- повышенная чувствительность и ранимость кожи и слизистых оболочек;
- появление пузырей, которые проходят без образования рубцов или с рубцеванием;
- образование грануляционной ткани — соединительной ткани, которая образуется на месте повреждений при их заживлении;
- повреждение зубной эмали.
Выделяют два подтипа данной формы:
- Подтип Херлитца, которому свойственно:
- большое количество пузырей;
- наличие эрозий и рубцов;
- заболевание или полное отсутствие ногтевых пластин;
- милии;
- тяжелые заболевания ротовой полости — кариес, воспаление мягких тканей, нарушение эмалевого покрытия зубов.
Дополнительными симптомами запущенной стадии могут быть анемия, пневмония, сепсис, заболевания глаз, нарушения работы желудочно-кишечного тракта, очаги грануляционной ткани на лице и в области подмышек.
- Подтип Не Херлитца, его симптомами могут быть:
- очаги пузырей и эрозии на коже;
- образование корок и рубцов;
- заболевания ногтей;
- кариес и нарушение эмали.
Симптомы обоих подтипов похожи, но вид Херлитца отличается более тяжелыми поражениями организма.
Синдром Киндлера
Отдельный вид, — при котором появление пузырей возможно на различных уровнях слоев кожи верхних и нижних конечностей. Их образование происходит уже к моменту рождения малыша. Помимо образования пузырей возможны следующие симптомы:
- развитие кариеса, пародонтита и других заболеваний полости рта;
- нарушение работы желудочно-кишечного тракта;
- развитие заболеваний глаз;
- дистрофия ногтей;
- появление проблем в функционировании мочеполовой системы.
По мере роста пациента количество вновь образующихся пузырей снижается, но при этом кожа становится очень тонкой, чувствительной, возможно появление мелких кровеносных сосудов, располагающихся близко к поверхности кожи.
Своевременная диагностика
Медицинское лечение при данном заболевании начинается с диагностики — она дает возможность установить точный диагноз, включая подтип эпидермолиза, что дает возможность назначения конкретного лечения. Помимо внешнего осмотра проводится ряд специфических исследований:
- иммунофлюоресценция;
- иммуногистология;
Они позволяют оценить уровень нарушения структуры генов и глубину затронутых болезнью слоев кожи.
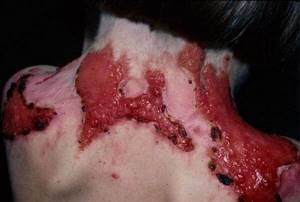
Также проводятся:
- биопсия;
- генетический анализ;
- микроскопия — для оценки состояния внутренних слоев кожи.
Обязателен сбор анамнеза пациента — получение информации о наличии семейных заболеваний, длительности болезни, ее проявлениях и симптомах, и комплексное обследование всех систем организма, анализы мочи и крови.
Своевременная диагностика и определение точного диагноза — подтипа буллезного эпидермолиза может дать возможность для улучшения качества жизни маленького пациента и создания препятствия для развития рецидивов заболевания.
Методы лечения буллезного эпидермолиза
Как правило, назначается комплексное систематическое лечение, которое может включать в себя следующие составляющие:
- Гигиенические процедуры пузырей, ран, очагов эрозии и язв на коже. Это необходимо для их скорейшего зарастания и уменьшения вероятности их разрастания — в качестве повязок чаще всего используют специальные не прилипающие бинты без вылезающих ниточек, способных травмировать нежную кожу пациента.
- Протеиновая терапия — требующая введения в организм некоторого количества белка.
- Клеточная терапия, при которой в организм пациента вводятся клетки, содержащие здоровые гены, отвечающие за кодирование белка.
- Генная терапия, характеризующаяся в организм генов, заменяющих таковые с нарушенной структурой.
- Некоторые методы комбинированного лечения.
- Применение для лечения стволовых клеток мозга, как донорских, так и собственных.
- Использование специализированных лекарственных препаратов, препятствующих блокировке кодирования белка.
- Лечение сопутствующих заболеваний — в зависимости от их тяжести и степени поражения организма.
Нужно отметить, что в настоящее время ведется активная работа по научным разработкам методов лечения буллезного эпидермолиза, как в Российской Федерации, так и за рубежом. Но единого способа диагностики и лечения данной болезни пока нет, а квалификация специалистов не везде на должном уровне, что не дает гарантии на благополучный исход для пациентов с подобным диагнозом.
Продолжительность жизни с буллезным эпидермолизом
Исход болезни и продолжительность жизни с диагнозом буллезного эпидермолиза напрямую зависит от степени изменения структуры генов и глубины поражения тканей и всего организма в целом. На состояние пациента влияет также подтип эпидермолиза.
Простые формы могут протекать относительно легче более тяжелых, сопровождающихся образованием большого количества пузырей, развитием инфекционного процесса и нарушением функций внутренних органов и систем организма.
Второй вариант течения болезни часто заканчивается летальным исходом еще в младенческом возрасте.
Продолжительность жизни людей с таким диагнозом, как правило, невысока. Помимо медицинских показаний, она зависит от степени ухода за больным — в данном случае родителям нужно приложить максимум усилий и терпения — это даст возможность улучшить качество жизни маленького пациента.
В медицинской истории описаны случаи так называемого обратного мозаицизма — когда при унаследованной мутации, изменяющей структуру определенных генов, происходит их восстановление вследствие работы определенных систем организма и новых изменений на генном уровне. Этот процесс крайне редкий, но, все же, такое чудо имеет место быть.
Каким бы неприятным ни казался диагноз, современная медицина может творить чудеса.
Своевременная диагностика, грамотное лечение и регулярные обследования могут дать возможность для благополучного исхода болезни или некоторого улучшения качества жизни пациента.
Конечно, результат в немалой степени зависит от тяжести болезни и общего состояния здоровья пациента, а в данном случае, к сожалению, генетические предрасположенности изменить невозможно.
Буллезный эпидермолиз
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие.
Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии.
Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи.
Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу.
Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Буллезный эпидермолиз
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику.
Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа.
При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки.
При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других.
Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332.
Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи.
Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем.
На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи.
В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы.
Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы.
Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные.
В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз – имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз – делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз – имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым.
Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии.
Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления.
При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается.
Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела.
Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции.
Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни.
У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы.
Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела.
Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы.
Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей.
Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных.
В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний.
Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза.
При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания.
На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы.
Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении.
Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза.
Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой.
Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон.
Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей.
При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения.
Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии.
Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту.
Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи.
Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Буллезный эпидермолиз: причины, классификация, симптомы, основные методы диагностики и лечение
Иногда в литературе можно встретить диагноз «механобуллезная болезнь». Но в справочниках дерматологии ее нет.
На самом деле речь идет о буллезном эпидермолизе – гетерогенной группе генетических патологий, обусловленных мутациями определенных генов, которые ответственны за синтез структурных белков кожи.
Их основной признак — появление на коже и слизистых пузырей с серозным содержимым при малейшей механической травме. В последующем на месте буллезной сыпи образуются долго незаживающие эрозии.
Классификация
В зависимости от глубины поражения различают три основные формы заболевания и более 30 подтипов патологии, отличающихся по генотипу, фенотипу и характеру наследования.
- Простая форма (ПБЭ). Затронуты только верхние слои эпидермиса. Имеет 12 подтипов, наиболее часто встречающиеся — синдромы Вебера-Коккейна (локализованный), Кёбнера (генерализованный), Доулинга-Меары (герпетиформный), более редкие – ПБЭ Огна, ПБЭ с мышечной дистрофией, ПБЭ с пятнистой пигментацией.
- Пограничная форма (ПгБЭ). Дно пузыря расположено на уровне светлой пластинки – поверхностном слое базальной мембраны. Имеет 2 подтипа. Один из них представлен 6 клиническими формами. Наиболее тяжелый тип, который отличается высокой смертностью – подтип Херлитца.
- Дистрофическая форма (ДБЭ) – страдает верхняя часть сосочкового слоя дермы, которая расположена глубже светлой пластинки. Также имеет два подтипа, которые различаются по механизму наследования – доминантный и рецессивный. Последняя разновидность представлена несколькими клиническими формами, наиболее тяжелая из них – подтип Аллопо-Сименса.
- Синдром Киндлера (смешанный буллезный эпидермолиз). Пузыри могут одновременно затрагивать разные уровни кожи. Самая редкая и мало изученная форма заболевания.
В настоящее время эта классификация считается условной, поскольку на практике встречается очень много клинических форм, из-за чего часто возникают трудности с диагностикой. Специалисты стараются разработать более структурированную и приемлемую классификацию болезни, но пока безуспешно.
Причина
Заболевание развивается вследствие мутации в более чем десяти генах, в которых закодированы белки, составляющие структуру кожи. В большинстве случаев оно передается по наследству, реже является приобретенным, когда изменения в генах происходят спонтанно.
Ребенок может унаследовать патологию от одного из родителей. И даже если они не страдают буллезным эпидермолизом, взрослые могут быть скрытыми носителями мутировавших генов.
Условно все причины разделяют на две группы – генетические и внутриутробные. Основной патогенез – сбой в ферментной системе, в результате чего некоторые протеины становятся мишенью для атаки ферментов.
Генетические нарушения очень разнообразны:
- мутации в генах PLEC, KRT5 и KRT14 вызывают простую форму заболевания, атаке подвергаются протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1;
- мутации в генах LAMB3, LAMA3, LAMC2 провоцируют развитие пограничной формы патологии, мишенью становятся коллаген 17-го типа и ламинин-332;
- мутации в гене COL7A1 – причина дистрофической формы, при них происходит повреждение коллагена 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи;
- мишенью при синдроме Киндлера выступает белок киндлин-1.
Образование пузырей происходит в результате деструкции ключевых структурных белков и нарушения связи между клетками при малейшем механическом воздействии.
Внутриутробные причины, которые могут привести к мутации генов – самые банальные – курение, употребление алкоголя, прием лекарственных препаратов и воздействие других факторов, обладающих тератогенным эффектом.
Формы заболевания и его симптомы
Кожа пациентов не терпит никакого грубого механического или физического воздействия. Иначе в ответ быстро наступает отторжение эпидермиса и дермы. Сначала образуются пузыри, после вскрытия на их месте появляются эрозии. В некоторых случаях к высыпаниям может присоединяться вторичная инфекция.
Люди с диагнозом буллезный эпидермолиз находятся в группе риска по возникновению злокачественных новообразований.
Врожденный буллезный эпидермолиз может проявляться уже при рождении. В некоторых случаях кожа ребенка травмируется даже в момент прохождения по родовым путям матери.
Малышей, родившихся с такой патологией, называют «дети бабочки».
Однако первые симптомы могут появиться и значительно позже – точных сроков нет. Это может быть период новорожденности, младенчества или раннего детства. Реже всего заболевание начинается в юношестве, но такие случаи также зарегистрированы.
Приобретенный
Приобретенный буллезный эпидермолиз, как уже было сказано выше, возникает в результате спонтанных мутаций. Поэтому чаще всего первые признаки появляются уже у взрослых людей.
Пузыри и эрозии на коже пациентов, страдающих буллезным эпидермолизом, напоминают ожоги 3 степени.
Клиническая картина во многом схожа с симптомами этой болезни у детей и зависит исключительно от того, в каких генах произошли изменения.
Простой буллезный эпидермолиз
- ПБЭ подтипа Доулинга-Меара (герпетиформный). Наиболее тяжелая форма патологии. Первые признаки видны уже при рождении. Высыпания генерализованные, иногда образуют большие эрозии. Чаще всего поражается слизистая ротовой полости. Патология сопровождается герпетиформными высыпаниями на теле и конечностях. При этом подтипе могут страдать ногти. Происходит их отторжение. Впоследствии отрастают деформированные ногтевые пластинки или длинные ногти с гиперкератозом. Его проявления также возникают на ладонях и подошвах, позже они переходят в кератодермию.
Простой герпетиформный БЭ (Доулинг-Меара) - ПБЭ Кебнера (генерализованный подтип). Обширные высыпания появляются при рождении. Их излюбленная локализация – кисти и стопы. После заживления эрозий остаются участки депигментации. Иногда возникает ладонно-подошвенный гиперкератоз.
ПБЭ тяжелый генерализованный (Кебнера) - ПБЭ подтипа Вебера-Коккейна (локализованный). Наиболее легкая форма. Впервые заболевание проявляется в младенчестве, реже – в детстве или юношестве. Излюбленная локализация пузырей этого подтипа – ладони и стопы, реже – волосистая часть головы. Незначительные эрозии возникают в ротовой полости. Часто при патологии появляется гипергидроз ладоней и подошв. После заживления эрозий остается пигментация.
ПБЭ локализованный (Вебера-Коккейна) - ПБЭ Огна. Характеризуется сезонным образованием пузырей. Высыпания появляются летом, преимущественно на конечностях. Они сопровождаются онихогрифозом (искривлением ногтя в виде когтя) на больших пальцах ног.
- ПБЭ с мышечной дистрофией. Появление генерализованных пузырей при рождении. Сопровождается прогрессирующей мышечной дистрофией.
- ПБЭ с пятнистой пигментацией. Обширные участки с гиперпигментацией на туловище, кистях рук и стопах. При этом пузырей образуется немного, но по мере их регресса пигментация может усиливаться. Дополнительным симптомом этого подтипа являются веррукозные папулы – бородавчатые наросты на коже. Слизистая ротовой полости страдает незначительно.
- ПгБЭ Гертлица. Самая тяжелая форма буллезного эпидермолиза. Она приводит к летальному исходу в младенчестве или в раннем детском возрасте. Первые проявления – это обширные пузыри, которые возникают при рождении ребенка. Позже появляются участки гипертрофии грануляционной ткани, преимущественно в области рта, глаз, ноздрей, волосистой части головы и ушных раковин. Дополнительный симптом этого подтипа – утрата ногтевых пластинок и образование гипертрофической грануляционной ткани на концевых фалангах. Страдает ротовая полость – ямки в зубной эмали, обширные эрозии на слизистой ротоглотки. Возможно поражение эпителия носовой полости, слизистой конъюнктивы, пищевода, трахеи, гортани, прямой кишки и уретры. В тяжелых случаях развиваются многочисленные системные нарушения. Заболевание протекает на фоне анемии, происходит задержка роста и физического развития. К летальному исходу приводит осложнение патологии — сепсис.
Пограничный генерализованный тяжелый ВБЭ (летальный Херлитца) - ПгБЭ не-Гертлица. Высыпания в ротовой полости и поражение дыхательных путей не столь тяжелое, как при предыдущем подтипе. Однако встречаются тяжелые аномалии эпителиальной адгезии («склеивания»), что требует проведения трахеотомии с последующей установкой трахеостомы. Наиболее распространенные симптомы – участки сыпи на волосистой части головы и дистрофия ногтевых пластинок, а также периорифициальные (расположенные вокруг естественных отверстий организма, чаще всего – вокруг ануса) эрозии.
ПогрБЭ генерализованный среднетяжелый (не Херлитца) - Локализованный ПгБЭ (минимальный). Течение заболевания довольно легкое. Высыпания локализуются на кистях, стопах и передней поверхности голени. Реже развивается дистрофия ногтей, и появляются небольшие углубления на зубной эмали. На слизистой ротовой полости и носа образуются мелкие эрозии. Прогноз благоприятный.
- ПгБЭ с атрезией (отсутствием или заращением) привратника. Еще одна тяжелая форма заболевания. Пациенты страдают от неимоверной хрупкости кожных покровов и слизистых. У них часто обнаруживают аномалии развития органов мочевыделения, например, гидронефроз или нефрит. Отличительная особенность – рудиментарные ушные раковины.
- Локализованный доминантный ДБЭ (ДДБЭ, подтип Коккейна-Турена). Пузыри образуются на участках, которые чаще всего подвержены трению – колени, область крестца и акральные поверхности. Впоследствии на месте сыпи кожа подвергается дистрофическим процессам, рубцеванию с образованием милиумов. Страдают ногти, вплоть до полного отторжения и атрофического рубцевания последних фаланг пальцев.
- Генерализованный доминантный ДБЭ (ДДБЭ, подтип Пазини). Течение патологии более тяжелое, чем у локализованной формы. Пузыри сменяются эрозиями. После их заживления образуются рубцовые бляшки и милиумы. Дополнительный симптом – спонтанное появление на теле бесцветных плотных папул. По мере взросления пациента генерализованные очаги регрессируют в локализованные с преимущественным поражением кожи конечностей. Заболевание сопровождается дистрофическим изменением ногтей, часто приводящим к утрате ногтевой пластинки, и образованием небольших эрозий на слизистой ротовой полости.
- Рецессивный ДБЭ (РДБЭ). Локализованная форма отличается средней степенью тяжести и по симптомам схожа с ДДБЭ Коккейна-Турена. Подтип Аллопо-Сименса – РДБЭ с тяжелым течением. При рождении возникают обширные генерализованные высыпания. Иногда у ребенка развивается масштабное отслоение кожи (синдром Барта). Чередование обострений и периодов нестойкой ремиссии приводит к распространенному рубцеванию, в результате чего часто образуются сгибательные контрактуры. Заболевание сопровождается проявлением псевдосиндактилии – пальцы ребенка находятся в своеобразной «варежке», что может привести к их сращению. Периорифицивального поражения нет, очаги высыпаний локализуются на голове и шее. Особенно страдает волосистая часть головы, где быстро прогрессирует алопеция. Одно из самых серьезных проявлений – обширное поражение слизистой ротоглотки. После эрозий образуются рубцы, которые ограничивают движения языка и мешают нормально открывать рот. На зубной эмали появляются точечные уплотнения, часто приводящие к кариесу и даже полной потере зубов. Очаги патологии на слизистой верхних дыхательных путей провоцируют стеноз, из-за чего пациенту часто приходится ставить трахеостому. Эрозии в пищеводе вызывают образование стриктур. Сочетание всех этих проявлений приводит к недоеданию и как следствие – задержке роста. У таких детей часто развивается анемия.
Рецессивный дистрофический тяжелый генерализованный БЭ (Аллопо-Сименса)
Наиболее часто у пациентов с РДБЭ, которые достигли полового созревания, развивается тяжелое осложнение – плоскоклеточная карцинома. Эта патология отличается агрессивным течением, склонностью к инвазии и быстрому метастазированию.
Первые признаки видны уже при рождении. Поскольку высыпания одновременно затрагивают различные ультрастуктурные уровни, клинические проявления могут напоминать как дистрофическую, так и пограничную форму буллезного эпидермолиза. В процессе заживления эрозий на коже происходят атрофические изменения.
По мере взросления буллезные высыпания проходят, но развивается пойкилодермия. Это проявления атрофии, гиперкератоза, сосудистых и пигментных нарушений на коже открытых участков тела, то есть тех, которые подвержены инсоляции. И чаще всего они возникают у пациентов с фоточувствительностью. Помимо этого патология затрагивает ногти и может привести к синдактилии пальцев рук и ног.
Со временем течение заболевания осложняется нарушениями со стороны внутренних органов. Возникают воспалительные процессы на слизистой ротовой полости и стриктуры в пищеводе и уретре.
Диагностика
Анамнез и клиническая картина позволяют установить только предварительный диагноз. Окончательно подтвердить буллезный эпидермолиз могут только методы лабораторной диагностики. Для этого проводят биопсию и образцы кожи подвергают ряду специфических тестов.
- Трансмиссионная электронная микроскопия. Помогает увидеть и оценить определенные элементы кожи, а также обнаружить в ней ультраструктурные изменения.
- Иммунофлюорисцентное антигенное картирование (ИАК). Определяет уровень образования пузыря. Позволяет оценить количество структурных белков и определить, какой из них стал мишенью, а также уточнить ферментную активность ткани. Уменьшение уровня ключевого протеина может свидетельствовать о его низком синтезе или массовом разрушении.
- Генетический анализ (молекулярная или ДНК-диагностика). Завершающий анализ. Прямое секвенирование (определение последовательности соединения нуклеотидов или аминокислот в цепи ДНК или РНК) дает возможность точно определить форму буллезного эпидермолиза и его тип наследования.
Если в семье есть родственники, страдающие буллезным эпидермолизом, целесообразно пройти генетическую консультацию на стадии планирования беременности, а также пренатальную диагностику, которая позволит выявить патологию на раннем этапе развития плода.
Методы лечения
- Буллезный эпидермолиз – системная патология, поэтому лечением занимается мультидисциплинарная команда врачей.
- Заболевание полностью излечить невозможно, поскольку специфические препараты для борьбы с ним до сих пор не найдены.
- Однако современные методы симптоматической терапии, правильный уход за ранами, а также превентивные меры значительно облегчают состояние пациентов и дают им возможность прожить довольно долго.
- Цель всех лечебных мероприятий:
- предупреждение разрастания и вскрытия пузырей;
- быстрое заживление эрозий и эпителизация кожи;
- предупреждение развития осложнений.
В качестве симптоматической терапии могут быть назначены следующие группы препаратов:
- анальгетики устранят боль;
- антигистаминные избавят от зуда и жжения;
- антибиотики помогут при вторичном инфицировании эрозий;
- глюкокортикоиды снимут воспаление и неприятные ощущения на коже;
- поливитамины окажут общеукрепляющее действие.
Наружное лечение и уход за кожей заключается в обработке пузырей антисептиками, наложении повязок, в основе которых неприлипающие материалы и несколько слоев бинтов. Кроме того, проводят УФО – профилактическое облучение кожи ультрафиолетовыми лучами для предупреждения инфицирования эрозий.
Продолжительность жизни с буллезным эпидермолизом
Прогноз и продолжительность жизни больного буллезным эпидермолизом зависит от многих факторов — формы патологии и ее подтипа, качества ухода, наличия осложнений и общего состояния пациента.
Раньше летальный исход наступал в младенчестве, реже малыши доживали до трех лет.
Простой буллезный эпидермолиз и некоторые локализованные формы других типов имеют наиболее благоприятный прогноз. Иногда проявления болезни регрессируют, и пациенты проживают обычную жизнь. Добиваясь стойкой ремиссии, они никогда не забывают о мерах профилактики и соблюдают все рекомендации врачей.
Наиболее неблагоприятный прогноз при дистрофической форме заболевания. Множественные осложнения часто приводят к летальному исходу еще в детстве.
В настоящее время, к сожалению, не все родители могут обеспечить ребенку-бабочке надлежащий уход. Однако специальные благотворительные фонды не оставляют малышей и помогают им по мере возможности.
Успешное лечение буллезного эпидермолиза
В 2015 году семилетний мальчик был госпитализирован с буквально слезающей кожей и волдырями на руках и ногах, а также на спине. Пациента спасали врачи из Германии, Австрии и Италии. По их словам, состояние мальчика было критическим: он потерял порядка 60% кожных покровов.
524 • • 30.11.2017
Специалисты поместили юного пациента в индуцированную (медикаментозную) кому, чтобы избавить от дальнейших страданий и начали с традиционной пересадки. Мальчику трансплантировали кожу отца ребёнка, а также донорские материалы. Но это не помогло.
Тогда родители согласились на экспериментальное лечение, которое провела команда из Рурского университета в Бохуме (Германия). Учёные использовали методы генной терапии, чтобы создать для маленького пациента новую кожу.
С тех пор прошло два года, и мальчик уже вернулся к обычной жизни: он ходит в школу, общается со сверстниками и даже играет в футбол. При этом ему больше не нужны лекарства или какое-то дополнительное лечение.
Генная терапия и раньше применялась для создания кожи, которая использовалась при пересадках. Однако в данном случае у пациента наблюдалась редкое и неизлечимое генетическое заболевание – буллезный эпидермолиз.
Отличительным признаком болезни является повышенная чувствительность кожи: она попросту не переносит механических воздействий. На коже постоянно появляются пузыри и волдыри, а также эрозии, язвы и открытые раны. В итоге кожа непрерывно шелушится и отпадает, а риск занести в раны инфекции невероятно высок.
В таком состоянии пациенты очень тяжело переносят операции по пересадке кожи. К тому же последние являются не очень-то эффективными, поскольку БЭ, как упоминалось выше, болезнь генетическая.
У человека наблюдаются мутации в генах, которые кодируют белки, отвечающие за состояние эпидермиса – по сути, за «закрепление» верхних слоёв кожи.
Когда эти белки производятся в недостаточном количестве или совсем отсутствуют, кожа становится настолько «хрупкой», что верхняя её часть слезает при малейшем воздействии. И пересадка новой кожи ничего не меняет: даже когда она станет частью организма, то точно так же начнёт разрушаться.
Буллёзный эпидермолиз проявляется в детстве, часто в младенчестве, и не только доставляет страдания детям, но и является прямой угрозой для жизни.
Некоторые пациенты не доживают до совершеннолетия, у других со временем развивается рак кожи. Одним словом, встретить взрослого человека с БЭ почти невозможно.
Медики и волонтёры называют юных пациентов с этим заболеванием «детьми-бабочками», проводя аналогию с тем, как насекомые сбрасывают несколько слоёв кокона.
Пересадка кожи или постоянное залечивание ран – единственный выход для пациентов с буллёзным эпидермолизом. По крайней мере, так было до появления нового метода лечения.
В ходе работы учёные взяли у пациента небольшой кусочек кожи из неповреждённой области. Из него извлекли стволовые клетки, которые заразили ретровирусом со здоровой копией гена под названием LAMB3 – того самого, мутации в котором и вызывают болезнь.
Затем из этих стволовых клеток исследователи вырастили крупные «листы» кожи (размером от 50 до 150 квадратных сантиметров) тем же способом, как выращивается кожа для пересадки после ожогов. В общей сложности специалисты вырастили около одного квадратного метра новой кожи. В ходе трёх операций её пересадили ребёнку, в конечном счёте заменив 80% его кожного покрова.
Спустя десять дней новая кожа начала «приживаться», запустились процессы регенерации, а через восемь месяцев. Почти вся кожа мальчика полностью восстановилась до нормального состояния: на ней появились потовые и сальные железы, начали расти волосы.
До сих пор никаких осложнений или побочных эффектов выявлено не было, отмечают эксперты. Они поясняют: в данном случае важно, чтобы иммунная система организма не посчитала новый ген чужеродным и не начала отторгать и атаковать клетки с «подменой».