Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.
мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Известные люди с синдромом Марфана
Синдром Марфана назван по имени педиатра, который наблюдал девочку с этим заболеванием на протяжении 20 лет. Имеется много интересных фактов о людях, имеющих характерные признаки патологии. Первая манекенщица (Лесли Хорнби – «Твигги»), которая была прототипом для всех чрезмерно худых моделей, болела синдромом Марфана. Наиболее известные личности, о которых есть подобные сведения:
- президент А. Линкольн,
- скрипач Н. Паганини,
- писатель Г. Х. Андерсен,
- композитор С. Рахманинов,
- олимпийский чемпион М. Фелпс.
Распространенность симптомокомплекса у многих одаренных людей дала возможность предположить, что он придает экстраординарные способности.
Рекомендуем прочитать статью об аортальном пороке сердца. Из нее вы узнаете о распространенности заболевания и причинах его развития, симптомах патологии, диагностике и лечении.
А здесь подробнее о симптомах аневризмы аорты.
К какому врачу обратиться
При подозрении на наличие синдрома Марфана следует обратиться к врачу-генетику. После проведения диагностики к наблюдению и лечению пациента подключаются и другие специалисты: кардиолог, ортопед, офтальмолог и терапевт. При необходимости проведения корригирующих операций на сердце и сосудах больного направляют к кардиохирургу.
Синдром Марфана является редким генетическим заболеванием, которое сопровождается неправильным развитием соединительной ткани. Впоследствии у больного возникают поражения скелетных структур, сердца и сосудов, органов зрения, нервной и других систем организма. Постоянное врачебное наблюдение за такими пациентами, регулярное диагностическое обследование и своевременное лечение возникающих патологий позволяют существенно улучшать качество жизни таких людей и предупреждают развитие жизнеугрожающих осложнений.
Медицинская анимация о симптомах синдрома Марфана (англ. яз.):
О синдроме Марфана в программе «Жить здорово!» с Еленой Малышевой (см. с 30:20 мин.):
Рейтинг: (голосов — 1, среднее: 5,00 из 5)
Причины заболевания
Болезнь связана с генетической аномалией, которая передается по наследству или возникает спонтанно. Мутациям подвергается часть хромосомы, которая отвечает за образование фибриллина. Этот белок делает соединительную ткань прочной и упругой. При его дефиците страдают структуры, которые имеют наибольшее количество подобных волокон:
- сосуды, особенно аорта;
- сухожилия и связки, в том числе хрусталика глаза;
- подкожная клетчатка.
Так как изменения генов могут быть разнообразными (изучено более 100 вариантов), а также они комбинируются с другими патологиями строения хромосомного аппарата, то проявлена синдрома Марфана отличаются.
О возможности лечения
Специфического лечения синдрома Марфана на сегодняшний день не существует. Но новые препараты и технологии помогают продлить жизнь пациента. Лечение при данном заболевании симптоматическое, то есть проводится при проявлении тех или иных симптомов. Важнейшим компонентом лечения являются регулярные осмотры врачей, прежде всего, кардиолога для предотвращения развития аневризмы аорты и её расслоения. Для реализации данной цели используются препараты, уменьшающие частоту сердечных сокращений (группа бета-блокаторов), а также лекарственные средства для снижения кровяного давления (ингибиторы ангиотензин-превращающего фермента). Если наблюдается расслоение аорты или аневризма, то резко повышается риск разрыва аорты и летального исхода. В такой ситуации требуется хирургическая коррекция аорты. Благодаря таким операциям, существует вероятность продлить жизнь пациенту.
Нарушение деятельности скелетной мускулатуры и костной системы также поддаётся лечению. При болевом синдроме используют болеутоляющие средства и миорелаксанты. Данные изменения, характерные для синдрома Марфана, имеют хороший результат после физиотерапевтического лечения и курса массажа.
При развитии спонтанного пневмоторакса пациенту необходима срочная помощь и госпитализация в торакальное отделение хирургического стационара.
Для коррекции зрения рекомендуется ношение корректирующих очков. Если произошло развитие глаукомы, то необходимо оперативное лечение.
Классификация заболевания
Встречаются легкие случаи и крайне тяжелые, с быстрой прогрессией нарушения деятельности сердечно-сосудистой, легочной системы, почек. Промежуточное положение между ними занимает синдром Марфана средней тяжести.
Также существует подразделение этого заболевания на клинические формы по наличию поражения систем организма:
- стертая – патология в одной или двух системах со слабыми отклонениями от нормы;
- выраженная – незначительные нарушения в 3 системах или серьезные в 1;
- тяжелая – прогрессирующее нарушение деятельности 3 систем.
Стабильным вариантом течения считается болезнь при наличии изменений в работе или строении органов, которые не прогрессируют на протяжении более 1 года.
Что это такое?
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью.
В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Наиболее опасными являются не внешние дефекты, придающие пациентам характерный вид, а патология строения сердца и сосудов. Именно эти пороки развития определяют продолжительность жизни больных и занимают ведущее место в клинической картине. Наиболее типичные из них:
- стеноз магистральных сосудов – аорты и легочной артерии;
- отверстия в перегородках сердца;
- аортальная недостаточность, расширение восходящего сегмента и аневризмы аорты;
- удлиненные створки митрального клапана с разрывом сухожилий, которые их прикрепляют.
Стеноз аорты при синдроме Марфана
Из-за поражений сосудов и строения клапанного аппарата пациенты страдают от недостаточности кровообращения, они подвержены развитию бактериального эндокардита, аритмии в виде тахикардии, мерцания или трепетания предсердий и желудочков. При комбинированных функциональных и анатомических нарушениях смертельный исход может быть на первом году жизни ребенка.
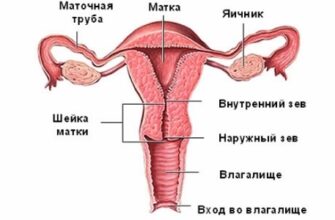
Нарушения зрения выявляются в виде двустороннего вывиха хрусталика, они, как правило, нарастают до 5 лет и сопровождаются потерей зрения. Также при синдроме Марфана возникают такие офтальмологические патологии:
- близорукость,
- увеличенная и более плоская роговица по сравнению с нормой,
- недоразвитая радужная оболочка,
- косоглазие,
- деформированные сосуды сетчатки.
При нарушении развития твердой оболочки мозга в поясничном отделе позвоночника возникает выпячивание содержимого спинного-мозгового канала, что сопровождается болью и онемением нижних конечностей. В легких возможно формирование эмфиземы, пневмоторакса, почки и мочевой пузырь, а у женщин и матка смещаются книзу, часто происходит растяжение связок, образование паховых и бедренных грыж.
По уровню умственного развития пациенты не отличаются от здоровых людей, а в некоторых случаях имеют очень высокий уровень интеллекта. Это связано с повышенным синтезом адреналина, поэтому такие люди излишне раздражительны, тревожны и возбуждены. Но при этом физическая нагрузка переносится плохо, сопровождается сильной мышечной и головной болью.
Смотрите на видео о синдроме Марфана:
Осложнения на сердце
В клинической картине этого заболевания часто доминирует сердечно-сосудистая патология. Она проявляется по-разному. У пациента могут наблюдаться стойкие дефекты стенок кровеносных сосудов. Они теряют свою эластичность. Особенно этому подвержены аорты, крупные ветви легочной артерии. У больных могут наблюдаться пороки развития клапанного аппарата и перегородок сердца.
Что касается аорты, то здесь чаще всего наблюдаются прогрессирующие расширения ее восходящей части и клапанного кольца (дилатация, аннулоаортальная эктазия). У больного часто имеются аневризмы, поражен митральный клапан. Наблюдается также патологическое удлинение створочных хорд и их разрыв.
У ребенка, который еще находится в утробе матери, но на генном уровне синдром Марфана ему уже передался, часто формируются врожденные пороки сердца. Это могут быть:
- коарктация аорт;
- дефект межпредсердной перегородки (ДМПП);
- стеноз легочной артерии;
- дефект межжелудочковой перегородки (ДМЖП).
Также может наблюдаться нарушение сердечного ритма (тахикардия, фибрилляция предсердий), развитие инфекционного эндокардита.
При самом неблагоприятном варианте неонатальной формы синдрома Марфана сердечная недостаточность быстро прогрессирует с самого рождения ребенка. Часто такие дети не доживают до одного года.
Диагностика заболевания
Синдром Марфана
Основные критерии, по которым можно поставить диагноз (хотя бы один из них):
- расширение или расслоение аорты,
- вывих хрусталика,
- грудная клетка с выпячиванием грудины,
- конечности, длиннее, чем в норме,
- ограничение разгибания локтевых суставов или деформация бедренных.
Если пациент сгибает кисть, сжимая большой палец, то его кончик выглядывает из кулака, длина среднего пальца превышает 10 см, а соотношение длины кисти в см и роста в метрах больше 11%, большим пальцем и мизинцем можно легко охватить запястье.
Данные инструментальных методов:
- ЭКГ – аритмия, гипертрофия сердечной мышцы.
- ЭхоКГ – расширение аорты, нарушения строения клапанов, повреждения хорд, увеличенный левый желудочек, видоизмененный митральный клапан.
- Рентген – аорта в восходящей части и сердце увеличены в размерах.
- КТ и МРТ подтверждают отклонения в строении магистральных сосудов и сердечных клапанов или стенок.
- Аортография выявляет признаки аневризмы и расслоения аортальных оболочек.
Показано обследование офтальмолога с биомикроскопией тканей глазного яблока, травматолога для диагностики нарушений функции суставов и позвоночника.
Подтверждение генетических мутаций проводится при помощи анализа ДНК. Типичным признаком синдрома Марфана является значительное превышение нормы выделения хондроитинсульфата и гиалуроновой кислоты с мочой.
Как ставится диагноз
Это заболевание в медицинской практике встречается нечасто. Как утверждают специалисты, на 10-20 тысяч человек только один может иметь синдром Марфана. Диагностика базируется на нескольких методах. Это семейный анамнез, а также исследования функций организма, офтальмологический осмотр, рентген и генетический прогноз. При этом нет разницы, какого пола человек, к какой расе он принадлежит и в каком месте Земного шара проживает. Это заболевание поражает в равной степени как жителей юга, так и севера.
Если доктор все же поставил диагноз «синдром Марфана», лечение может быть как консервативным, так и хирургическим. К оперативному вмешательству приходится прибегать в серьезных случаях, когда лечение с помощью медикаментов и физиологических процедур не дает нужных результатов.
Оперативному вмешательству чаще всего подвергают сердечно-сосудистую систему и органы зрения.
Если имеются значительные деформации скелета, то используют протезирование суставов, пластику грудной клетки, стабилизацию поясничного или шейного отдела позвоночника.
Прогноз и профилактические мероприятия
Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. При наличии серьезных нарушений со стороны этих органов длительность жизни пациентов ограничивается 40-50 годами, и повышается риск внезапной смерти. С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность.
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Правила жизни
Правила жизни при синдроме Марфана
Больные с синдромом Марфана должны соблюдать такие рекомендации:
- Низкий уровень физических нагрузок, запрет на соревнования, профессиональный спорт, а особенно борьбу, подводное плавание, тяжелую атлетику.
- Нельзя поднимать тяжести более 3 кг.
- Избегать вредных условий производственной деятельности, противопоказана работа, требующая нагрузку на зрение, тяжелый физический труд.
- Запрещено пребывание в условиях высокой радиации или температуры.
Для детей физкультуру можно проводить только в специальных группах в виде лечебной гимнастики.
Историческая справка
Первые упоминания о необычном недуге можно обнаружить в трудах американского офтальмолога Э. Вильямса, который в 1875 году описал признаки идентичного смещения хрусталиков глаз у родных брата и сестры. Кроме офтальмологических проблем эти дети имели повышенную подвижность суставов и высокий рост.
Известность болезнь приобрела позже, через 20 лет, когда французский педиатр Антуан Марфан представил свои наблюдения за 5-тилетей больной. Маленькая пациентка отличалась необычными аномалиями скелета и быстрым прогрессированием недуга. Синдром был назван в честь французского доктора, хотя впоследствии стало известно, что наблюдаемая им девочка страдала другой наследственной патологией – врождённой контрактурной арахнодактилией.
Можно найти множество примеров обнаружения синдрома у талантливых, знаменитых людей. Считается, что этим недугом страдали скрипач Никколо Паганини, американский президент Авраам Линкольн, русский композитор Сергей Рахманинов и другие известные личности. Некоторые исследователи считают, что неординарность людей с синдромом Марфана объясняется увеличенной концентрацией адреналина в крови. Этот гормон вызывает повышение активности и развитие незаурядных способностей.
Причины и симптомы патологии
На сегодняшний день было зафиксировано достаточно много проявлений этого заболевания. Синдром Марфана может быть практически незаметным и только в редких случаях приводит к инвалидности с детства.
Отметим, что люди с такой мутацией практически всегда имеют высокий рост – от двух метров. Один из основных симптомов – худое тело и длинные руки. Также выделяют необычно длинные пальцы. Синдром Марфана сопровождается дистрофией мышц, поэтому такие люди имеют астенический вид.
Признаки синдрома Марфана
В результате детальной диагностики синдрома Марфана были выделены дополнительные симптомы:
- Суставы гиперподвижны.
- Тазобедренная кость развивается аномально.
- Развитие кифоза или сколиоза.
- Частые вывихи шейного позвоночника.
- Нарушения в формировании грудной клетки.
- Плоскостопие.
- Глаза на лице посажены достаточно глубоко.
- Непропорционально маленький размер нижней челюсти.
- Небо расположено высоко.
- Частое формирование растяжек на коже.
- Образование грыж.
Поражения костей и зрения
Люди с болезнью Марфана чаще отличаются диспропорциональностью внешнего облика (отношение размаха рук к росту – 1,05). Они высокого роста, с непропорционально удлиненными конечностями и кистью с длинными пальцами (арахнодактилия). У пациентов развиваются патологии позвоночного столба – сколиозы, грудная клетка деформируется вовнутрь (воронкообразная) или наружу (килевидная).
У пациентов часто встречается нарушения зрения (астигматизм, близорукость, реже дальнозоркость). Эктопии хрусталика (аномалии расположения) регистрируются у 80% пациентов. Чаще патологии зрения проявляются после ослабления соединительных тканей у взрослых, но для пациентов с болезнью Марфана часто регистрируют глаукому в детском и юношеском возрасте.
Хирургические операции
При этом синдроме часто приходится прибегать к кардиохирургическим вмешательствам. Это позволяет существенно увеличить выживаемость больных. До применения операций на сердце продолжительность жизни пациентов с этим синдромом была небольшой. Люди доживали только до среднего возраста (40-50 лет). В настоящее время кардиохирургическое лечение увеличило жизнь пациентов примерно на 20 лет.
Нередко приходится делать операции на аорте. Они показаны при расширении этого сосуда более 5 см. Также осуществляют протезирование митрального клапана.
Операции на сердце делают чаще всего при таком синдроме, так как этот орган сильно страдает от дефектов соединительной ткани. Однако проводят и другие хирургические вмешательства, такие как:
- Торакопластика. Эту операцию делают при деформации грудной клетки. Она позволяет избавиться от дефекта строения скелета. Проводится частичная резекция нескольких ребер, после этого грудина выглядит более ровной.
- Экстракция хрусталика. Такую операцию на глазах проводят при ранней катаракте и глаукоме.
- Стабилизирование позвоночника с помощью металлических пластин. Операцию проводят в тяжелых случаях сколиоза.
- Эндопротезирование. У больных часто отмечаются патологии костей. По этой причине иногда приходится заменять пораженный сустав на протез.
- Удаление аденоидов и гланд. Эти вмешательства необходимо провести в детстве. Ребенок, страдающий данным синдромом, очень подвержен простудным заболеваниям.
Кроме этого, беременным женщинам с болезнью Марфана показана операция кесарева сечения. Из-за слабости мышц и проблем с сердцем им очень трудно рожать самостоятельно.
Самые серьезные последствия болезни
Нарушения в системе кровообращения и деятельности сердца являются наиболее серьезными осложнениями. Нарушение ритма сердечных сокращений, одышка и усталость, холодные конечности – вот внешние проявления, которые должны насторожить пациента. Наличие шумов в сердце.
Патологические проявления в электрокардиограмме, стенокардия являются основаниями для дальнейшего обследования. Нарушения в соединительных тканях сердца проявляются в пролапсе клапанов (митрального и аортального), расширенная аорта или ее аневризма при болезни Марфана (фото ниже).
Особую опасность представляет заболевание для женщин в период беременности. Даже при отсутствии видимых осложнений в период беременности у пациенток с таким диагнозом повышается риск расслоения аорты, что может привести к летальному исходу. Женщинам с таким диагнозом рекомендуется при планировании беременности пройти тщательное обследование сердечнососудистой системы, а в период вынашивания ребенка каждые шесть недель проходить эхокардиографическое исследование. Естественные роды у таких пациенток вполне возможны.
Ведущими критериями диагностики считаются нарушения в работе сердечно-сосудистой системы, особенно аневризма и расширение аорты. Но окончательный диагноз ставится при комплексном обследовании (существует более 30-ти клинических признаков заболевания) при участии генетиков, кардиологов, офтальмологов, ортопедов.
Если при исследовании семейной истории установлено, что болезнь наследственная, то диагноз подтверждается при наличии нарушений как минимум в работе двух систем организма. Если наследственный характер заболевания не установлен, то диагноз подтверждается при наличии поражения в трех системах.
Данная патология не имеет половых, этнических или социальных предпочтений. Большинство пациентов имеют родственников с данным заболеванием, но до 15% всех случаев связаны с новыми генными мутациями. Среди тех, кто имеет синдром Марфана, довольно много успешных людей как современности, так и прошлого.
Кроме особенностей скелета, эта болезнь награждает своих обладателей высоким уровнем адреналина в крови, повышенной работоспособностью и быстрыми реакциями, крутым нравом и жизнелюбием. Это болезнь, которая входит в пятерку самых распространенных среди знаменитостей. Приведем лишь несколько примеров успешных людей с данным синдромом.
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
В зависимости от характера течения, недуг может быть:
- стабильный;
- прогрессирующий.
Ссылки
На русском языке
- [dst.omsk-osma.ru/ «Дисплазия соединительной ткани»] — Медицинский информационный сайт (Омская государственная медицинская академия)
- [www.marfan.0pk.ru/ Российское интернет-сообщество людей, страдающих синдромом Марфана]
- [mma.ru/article/id36215 Синдром Марфана] — Портал ГОУ ВПО ММА им. И. М. Сеченова
На английском языке
- [marfanworld.org/ International Federation of Marfan Syndrome Organisations]
- [www.marfan.org/ National Marfan Foundation (USA)]
- [www.ncbi.nlm.nih.gov/disease/Marfan.html National Institute for Health Marfan syndrome page (USA)]
- [www.marfanssyndrome.net/ Marfan Syndrome Information & Support]
- [marfansyndrome.researchtoday.net/ Marfan Syndrome Research] — Recent literature on Marfan Syndrome
- [www.supportmarfan.com Marfan support]
- [heartcenter.seattlechildrens.org/conditions_treated/marfan_syndrome.asp Marfan Syndrome information] from Seattle Children’s Hospital Heart Center
- [www.marfan.ca/ Canadian Marfan Association]
- [www.marfan.org.uk/ Marfan Association UK]
- [www.marfan.org.mx/ Marfan de Mexico]
- [www.marfan.no/ Norwegian Marfan Organization]
- [www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=154700 Online Mendelian Inheritance in Man]
| В данной статье или разделе имеется список источников или внешних ссылок, но источники отдельных утверждений остаются неясными из-за отсутствия сносок. Утверждения, не подкреплённые источниками, могут быть поставлены под сомнение и удалены. Вы можете улучшить статью, внеся более точные указания на источники. |
Как наследуется патология?
Наследование синдрома Марфана происходит по аутосомно-доминантному типу. Если болен один родитель, то он может передать патологию ребенку в 50% случаев. Если же этим синдромом страдают и отец, и мать, то вероятность рождения больного малыша увеличивается до 75-100%.
Чаще всего патология передается от больных родителей. Но бывает и так, что отец и мать здоровы, однако ребенок страдает синдромом Марфана. Это отмечается в 15% случаев. Причины такой случайной мутации гена неизвестны. Однако специалистами установлено, что риск рождения ребенка с такой патологией увеличивается, если возраст отца превышает 35-40 лет. Возраст матери в этом случае не влияет на мутацию гена.
Другие проявления
У пациента появляются растяжки на коже, которые никак не связаны с изменением массы тела. Это является лишь косметическим недостатком и обычно не доставляет никакого беспокойства.
Однако пациенты очень склонны к образованию грыж в паховой и брюшной области, которые часто рецидивируют, несмотря на лечение.
Лечебная физкультура при болезни
Для того, чтобы добиться эффективного устранения этого заболевания следует использовать только допустимый уровень нагрузок, который будет способствовать улучшению физического состояния, набору мышечной массы. Это позволит аорте добиться нормального диаметра, а также нормализует работу органов дыхания
Ребенок сможет лучше концентрировать внимание, поможет усовершенствовать мелкую моторику рук. После проведения подобной терапии наблюдается улучшение успеваемости в школе
Требования, которые должны соблюдаться в режиме для людей, больных синдромом Марфана:
- Физическая нагрузка может производиться только по упрощенной программе. Лучше всего для этого посещать специальную реабилитационную группу.
- Ребенок не должен посещать спортивные секции, а также принимать активное участие в занятии спортом. Также следует устранить выполнение работ, которые связаны с большими физическими нагрузками. При передвижении не следует брать с собой вес более трех килограмм.
- Не следует выполнять работы, которые отличаются относительно вредным воздействием на организм человека. Следует полностью исключить контакт с различными химическими веществами такими, как краски, лаки. Негативным образом на состояние здоровья влияет работа при высоких температурах, радиацией. Не следует выполнять действия, которые требуют высокой концентрации зрения, а также производятся в нервной обстановке.
- Ребенок с данным синдромом не должен жить в жарком климате, а также в зоне с повышенной радиацией.
- Женщина, с синдромом Марфана, во время беременности должна один раз в два месяца проходить эхокардиографию. В том случае, если диаметр аорты будет более чем сорок пять миллиметров, то необходимо прерывать беременность искусственным путем, иначе жизни женщины будет угрожать серьезная опасность.
Клиника и синдромы
Синдром Марфана – болезнь соединительной ткани, и все проявления связаны с изменениями костей, суставов, кожи. Уникальных и однозначных симптомов у болезни нет. Но при наличии таких признаков, как удлиненные конечности и пальцы (паучья кисть), аномалии дислокации хрусталика и аневризма аорты вполне возможно предположить, что в генотипе пациента имеется аномальный ген болезни Марфана. Все синдромы заболевания делятся на:
- поражения костной системы;
- поражения системы кровообращения;
- поражения глаз;
- влияние на дыхательную систему;
- влияние на нервную систему.
Болезнь Марфана может проявляться в умеренной или тяжелой формах, все зависит от степени влияния на системы органов. При незначительном проявлении болезни все может ограничиться для ее носителя длинными руками и ногами с худыми пальцами. Чем страшна болезнь Марфана?