Одной из малораспространенных, но опасных болезней, является синдром Криглера-Найяра, способный привести к летальному исходу еще во младенческом возрасте. Патология напрямую связана с механизмами выведения билирубина из организма.
Заболевание может встречаться у пациентов всех возрастов и практически не поддается лечению. Чтобы избежать опасных осложнений, необходимо своевременно диагностировать и купировать синдром.
Общая характеристика заболевания
Среди всех хронических заболеваний печени, ухудшающих ее функциональность, выделяют врожденную гипербилирубинемию или синдром Криглера-Найяра, которая передается по наследству и встречается очень редко среди пациентов.
Механизм прогрессирования болезни основывается на дефиците ферментов, обеспечивающих обмен билирубина. Как правило, синдром проявляется как внешними признаками, так и устойчивыми неврологическими нарушениями.
- Полностью вылечить патологию пока невозможно, однако существует симптоматическое лечение, позволяющее привести показатели ферментов и билирубина к удовлетворительным и улучшить качество жизни пациентов.
Классификация
По количеству вырабатываемых ферментов выделяют синдром Криглера-Найяра 1 и 2 типа.
Для первого типа характерно:
- отсутствие глюкуронилтрансферазы как таковой;
- прогрессирование уже с первых дней жизни;
- тяжелое и болезненное течение;
- отсутствие возможности провести лечение, летальный исход в возрасте около двух лет.
Второй тип характеризуется признаками:
- отклонение уровня глюкуронилтрансферазы от нормы около 20% (дефицит);
- доминантный тип наследования (первому соответствует рецессивный);
- сравнительно более легкое течение;
- проявляется характерной симптоматикой в первый год после рождения;
- позволяет проводить симптоматическое лечение для поддержания жизни пациента в течение многих лет.
Таким образом, качество и результат лечения во многом будет зависеть от того, какой тип болезни диагностируется. В обоих случаях подход к терапии несколько различается.
В первом случае при помощи традиционных методов увеличить срок жизни удается, как правило, незначительно. Во втором случае возможно постоянное поддержание билирубина на удовлетворительном уровне для поддержания нормальной жизнедеятельности пациента.
Симптомы
Проявление патологии также зависит от типа синдрома Криглера-Найяра. Так, синдрому первого будут соответствовать следующие признаки:
- возникновение уже в первые часы после рождения;
- быстрое и внезапное пожелтение кожного покрова и слизистых;
- непроизвольное подергивание глаза (нистагм);
- гипертонус конечностей и шеи;
- отсутствие связанного билирубина в анализах желчи;
- внезапное повышение уровня билирубина и его устойчивость на высоком уровне (отклонение от нормы в 20-50 раз);
- постоянные судороги.

В первую фазу прогрессирования ребенок проявляет апатию и слабость, плохо сосет молоко, слабо дышит и имеет замедленную реакцию. При второй фазе нередко ребенок постоянно кричит, наблюдается избыточный тонус, судороги, набухание родничка.
Во время третьей фазы все болезненные признаки первых двух стадий практически полностью исчезают. Однако при четвертой набирают новую силу. В дальнейшем проявляется нистагм, отсутствие реакции, паралич. Смерть наступает уже через один-два года после рождения в 99% случаев. В редких случаях дети достигают подросткового возраста.
Признаки второго типа синдрома Криглера-Найяра:
- проявление только спустя некоторое время после рождения, в первые годы;
- пожелтение кожного покрова (как правило, только в подростковом возрасте);
- наличие излишнего тонуса в руках и ногах (некритично);
- судороги;
- в каловых массах можно обнаружить повышенный уровень уробилиногена.
Вероятно некоторое отставание в физическом и интеллектуальном развитии. Однако при этом продолжительность жизни практически не сокращается при правильно подобранном лечении.
Диагностика
При любом типе болезнь Криглера-Найяра определяется только после проведения первичного осмотра, а также лабораторных и аппаратных исследований. Пациентов любого возраста подвергают тестированию на реакцию, наличие судорог и других внешних проявлений. Далее проводятся следующие исследования:
- Биохимический анализ крови на определение уровня билирубина – многократное повышение последнего указывает на возможное наличие синдрома;
- Анализ генов – предполагает обнаружение гена, вызвавшего наследственное нарушение работы печени;
- Тестирование с использованием фенобарбитала – позволяет определить не только наличие заболевания, но и его тип: препарат не снижает уровень билирубина при первом типе болезни и снижает при втором;
- Биопсия органа – позволяет выявить наличие патологических включений в печени.
После постановки диагноза и определения типа синдрома, проводится подбор адекватного лечения.
Лечение
Терапевтическое воздействие в обоих случаях направлено на уменьшение и стабилизацию уровня билирубина в организме. Также нужно предотвратить токсические деструктивные изменения в головном мозге.
Первый тип
При синдроме Криглера-Найяра первого типа при помощи медикаментов и процедур продлить жизнь пациента и улучшить ее качество обычно удается незначительно. Для этого применяются:
- Введение в вену плазмы донора для выведения билирубина. Дозировка определяется в каждом конкретном случае индивидуально;
- Физиотерапевтическое воздействие для улучшения результата использования плазмы крови.
Альтернативным и наиболее сложным вариантом терапии является пересадка донорской печени. Этот метод позволяет существенно продлить жизнь пациенту, однако в настоящее время является трудноосуществимым.
Второй тип
Прогрессирование синдрома второго типа легче поддается терапии. В данном случае применяются методики:
- Введение плазмы.
- Физиотерапия для улучшения результатов лечения плазмой и ускорения процессов выведения билирубина из организма.
- Применение Фенобарбитала в дозировке от 1 до 5 миллиграммов на килограмм веса в сутки. Расчет ежедневной дозы зависит от возраста пациента.
- Плазмаферез. Способствует выведению билирубина, используется на усмотрение лечащего врача).

Дополнительно назначается активированный уголь для уменьшения воздействия токсинов на организм и головной мозг, а также для выведения билирубина из кишечника.
При такой терапии синдром Криглера-Найяра легко купируется и практически не проявляется в течение жизни. Периодически возможно кратковременное пожелтение кожи и склер глаз.
Осложнения и прогноз
К летальному исходу обычно приводит прогрессирующая энцефалопатия, наблюдающаяся при развитии болезни первого типа. Такой прогноз определяется в большинстве случаев, если пациенту не проводится пересадка печени.
При своевременной и правильно подобранной терапии синдрома Криглера-Найяра 2 типа осложнения не развивается, а прогноз благоприятный в большинстве случаев. В таких ситуациях продолжительность жизни практически не сокращается, а ее качество сохраняется на высоком уровне.
Профилактика
Предупредить возникновение заболевания генетического типа довольно сложно. Однако профилактические меры для семейных пар все-таки существуют.
Прежде всего, важно правильно планировать ребенка. Если хотя бы у одного из родителей имеется повышенная выработка билирубина или мутирующий ген, необходимо провести специальные тесты для определения вероятности рождения здорового ребенка.
Если риски окажутся слишком большими, семейной паре будет рекомендовано воздержаться от рождения ребенка.

Патология Криглера-Найяра второго типа характеризуется хроническим постоянным течением, которое включает в себя периоды ремиссии и обострения. Для того, чтобы предотвратить возникновение неприятных симптомов, необходимо вести здоровый образ жизни:
- Отказаться от всех вредных привычек. Особенно это касается принятия алкогольных напитков, серьезно влияющих на функционирование печени.
- Избегать эмоциональных потрясений и излишних моральных и физических нагрузок.
- Предотвращать и своевременно устранять все вирусные, грибковые и бактериальные инфекции, избегать простудных заболеваний.
- Отказаться от приема медикаментов, влияющих на функционирование печени.
При необходимости приема сильнодействующих лекарственных средств следует проконсультироваться с лечащим врачом о возможностях их использования и противопоказаниях. Любая интоксикация в организме может привести к резкому ухудшению самочувствия и ускорению прогрессирования синдрома Криглера-Найяра.

Заболевания, характеризующиеся аномально высоким уровнем выработки билирубина, опасны вероятностью повышенной интоксикации организма, способной привести к летальному исходу. Синдром Криглера-Найяра может купироваться консервативным лечением.
Однако многое в этом случае будет зависеть от типа заболевания и методов его лечения. Существенно улучшить прогноз при синдроме первого типа удается только после пересадки печени, что далеко не всегда оказывается возможным.
Лечение синдрома второго типа чаще всего приносит хорошие результаты и позволяет пациентам прожить долгую жизнь.
МКБ: E80.5 Синдром Криглера-Найяра: Расшифровка кода, лечение
Патогенез синдрома Криглера-Найяра
Синдром Криглера. Найяра — генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина — конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте.
Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками.
Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Синдром Криглера-Найяра – тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами – Джоном Криглером и Виктором Найяром.
Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии.
Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены – большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек.
В лечении этого состояния крайне важна ранняя (в идеале – пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
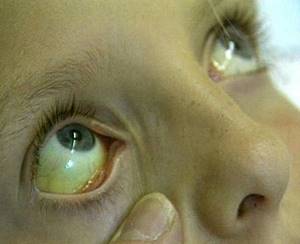 Синдром Криглера-Найяра характеризуется выраженной иктеричностью склер
Синдром Криглера-Найяра характеризуется выраженной иктеричностью склер
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации – к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты.
В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление.
Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи.
Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента.
Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера – синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1.
Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков.
Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением – первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения – нистагм, судорожные приступы, иногда возникает опистотонус.
Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе – участке, отвечающем за экспрессию данного гена.
У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой – промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность.
Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения.
В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе.
Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента.
С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов).
У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2.
Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение.
Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком – он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии.
Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС.
Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода.
Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора – в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови – все эти процедуры направлены на удаление неконъюгированного билирубина из организма.
Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия – облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови.
Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л – при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Синдром Криглера-Найяра
Синдром Криглера-Найяра – это хроническое злокачественное функциональное заболевание печени, передающееся по наследству и характеризующееся поражением нервной системы и выраженной желтухой. Описано это заболевание было в середине 20 века двумя учеными (Криглером и Найяром).
Данная патология поражает в основном жителей Азии (на 75-80%), встречается крайне редко, примерно 1 случай на 1 млн. новорожденных детей. В одинаковой степени поражает как мальчиков, так и девочек. Имеет достаточно высокую летальность (смертность), преимущественно в раннем возрасте.
Причины возникновения
Заболевания носит наследственный характер. Мутация, происходящая в некоторых генах, приводит к тому, что происходит нарушение выработки специфического фермента глюкуронилтрансферазы, который в норме связывает билирубин и трансформирует его в прямой (конъюгированный).
При нарушении выработки этого фермента, свободный (непрямой, неконъюгированный) билирубин, образующийся из гемоглобина в крови, попадает в печень, где не в состоянии связаться с ферментом, начинает оказывать свои токсические действия на печень, ЖКТ и нервную систему.
При этом желчь, выделяемая из печени, через желчный пузырь и желчные протоки, не содержит или практически не содержит билирубин.
Классификация
Существует два типа синдрома Криглера-Найяра:
- Первый тип – полное отсутствие фермента глюкуронилтрансферазы, аутосомно-рецессивный тип наследования, развивается с первых дней после рождения, имеет крайне тяжелое течение, заканчивается летально в первые 2 года жизни;
- Второй тип – отсутствие фермента глюкуронилтрансферазы на 20 % от нормы, аутосомно-доминантный тип наследования, носит относительно доброкачественный характер, первые признаки возникают в первые годы жизни ребенка, человек может прожить с данной патологией многие годы при адекватном выполнении всех рекомендаций врача.
Симптомы синдрома Криглера-Найяра
Симптомокомплекс при синдроме Криглера-Найяра зависит от типа заболевания.
Симптомы первого типа синдрома Криглера-Найяра
- Начало заболевания в первые часы-дни жизни ребенка;
- Резкая желтушность кожи и видимых слизистых (склеры, полость рта);
- Судороги;
- Повышенный тонус (гипертонус) на руках, ногах, шее;
- Нистагм (непроизвольное подергивание глазного яблока);
- Резкий подъем уровня билирубина – более 200 мкмоль/л, неконъюгированный билирубин выше нормы в 20-50 раз;
- Желчь не содержит связанный билирубин;
- Отставание в умственном развитии;
- Летальный исход возникает в первый-второй год жизни от билирубиновой энцефалопатии.
Билирубиновая энцефалопатия имеет 4 фазы развития и свои симптомы:
Первая фаза:
- Повышенная сонливость;
- Апатия;
- Ребенок слабо сосет;
- Мышечный гипотонус (все тело полностью расслаблено);
- Редкое дыхание;
- Монотонный плач, крик;
- Резкая реакция на минимальные раздражители;
- Рвота ранее съеденной пищей;
- Цианоз (синюшная кожа);
- Блуждающий взгляд.
Вторая фаза:
- Пронзительный, практически постоянный крик;
- Резко повышенный тонус в ножках и ручках (не разгибаются колени, ручки сжаты в кулачки);
- Ригидность затылочных мышц (невозможно наклонить головку ребенка так, чтобы он подбородком дотянулся до грудной клетки);
- Выбухание большого родничка;
- Выраженный тремор (дрожь) рук;
- Полное отсутствие сосательного рефлекса;
- Замедление частоты сердечных сокращений;
- Судороги;
- Замедление дыхания с периодами апноэ (отсутствие дыхания на короткое время);
- Нистагм.
Третья фаза:
- Кратковременный период ложного благополучия. Все признаки 2 фазы практически полностью уходят.
Четвертая фаза:
- Нистагм;
- Ребенок не умеет держать самостоятельно головку;
- Параличи и парезы тела;
- Отсутствие реакции ребенка на родителей;
- Отсутствие реакции ребенка на игрушки и другие отвлекающие предметы;
- Летальный исход, как финал заболевания (в 1% случае пациентов с 1 типом данного синдрома доживают до 10-15 лет, остальные погибают от 6 месяцев до 2 лет).
Симптомы второго типа синдрома Криглера-Найяра
- Первые признаки заболевания возникают в раннем детском возрасте (первые года жизни);
- Желтуха – появляется чаще всего в подростковом возрасте;
- Редкие судороги;
- Незначительное отставание в физическом и психическом развитии (данный симптом может и отсутствовать);
- Несколько повышенный тонус в конечностях;
- Повышение уровня билирубина и несвязанного билирубина (выше нормы в 5-20 раз);
- В кале повышенное содержание уробилиногена;
- Продолжительность жизни практически не уменьшается.
Диагностика
- Биохимический анализ крови – повышение общего и неконъюгированного билирубина (резкий подъем при 1 типе и умеренный при 2 типе) до 350-850 мкмоль/л и выше, отсутствие конъюгированного билирубина;
- Генетический анализ – обнаружение мутированного гена (UGTIAI);
- Проба с фенобарбиталом – при первом типе синдрома билирубин не снижается на прием фенобарбитала (проба отрицательная), при втором типе – билирубин снижается (проба положительная). Является патогномоничным признаком при различии типов синдрома Криглера-Найяра;
- Биопсия и последующая микроскопия печени – отсутствие пигментных включений. Патогномоничный отличительный признак от синдрома Дубина-Джонсона.
Лечение синдрома Криглера-Найяра
Терапевтические мероприятия первого и второго типа данного синдрома несколько отличаются друг от друга.
Лечение первого типа синдрома Криглера-Найяра
- Внутривенное введение донорской плазмы крови – происходит разрушение неконъюгированного билирубина и его выведение из организма. Кратность введения и количество плазмы подбирается индивидуально под каждого пациента;
- Фототерапия (метод физиотерапии) – способствует разрушению несвязанного билирубина и усиливает эффект от донорской плазмы;
- Трансплантация печени – способствует значительному улучшению обмена билирубина и продлевает жизнь таким пациентам на многие годы.
Лечение второго типа синдрома Криглера-Найяра
- Фототерапия (метод физиотерапии) – способствует разрушению несвязанного билирубина и усиливает эффект от донорской плазмы;
- Фенобарбитал принимается внутрь или вводится внутривенно (в зависимости от возраста и тяжести состояния пациента). Взрослым – по 1-5 мг на каждый кг веса, а детям – по 3-5 мг на каждый кг веса в сутки;
- Плазмаферез – проводится при обострении заболевания для ускоренного снижения несвязанного билирубина;
- Активированный уголь – 1 таблетка на каждые 10 кг веса пациента за прием, до 3 раз в сутки, между приемами пищи – позволяет сорбировать и вывести несвязанный билирубин из полости кишечника.
Осложнения
При первом типе синдрома Криглера-Найяра за 1-2 года от начала заболевания (зачастую с рождения) развивается билирубиновая энцефалопатия, которая приводить к летальному исходу.
Второй тип данного заболевания протекает без осложнений и его прогноз благоприятный.
Профилактика
В качестве профилактики рождения детей с синдромом Криглера-Найяра семейным парам, в анамнезе которых есть данная патология, необходимо пройти консультацию генетика и сдать кровь на пораженные гены. В случае уже имеющихся детей с такой болезнью рекомендовано отказаться от последующих родов.
При наличии синдрома Криглера-Найяра второго типа рекомендовано соблюдать несколько правил:
- Не употреблять гепатотоксичные медикаменты и средства, вытесняющие билирубин при связывании глюкуронилтрансферазу (гепарин, аспирин, оральные контрацептивы и пр.);
- Не употреблять алкогольные напитки;
- Избегать физических нагрузок, в особенности тяжелых;
- Избегать всевозможных инфекций и простудных заболеваний;
- Соблюдать все рекомендации лечащего врача;
- Своевременно проходить повторные обследования.
Синдром Криглера-Найяра
Синдром Криглера-Найяра (другое название – семейная желтуха) – наследственная аутосомно-рецессивная болезнь, характеризующаяся неконъюгированной злокачественной гипербилирубинемией (повышением уровня токсического билирубина в плазме крови). Проявляется тяжелыми неврологическими нарушениями и желтухой.
При этом виде гипербилирубинемии нарушается процесс конъюгации билирубина и глюкуроновой кислоты, проявляемый большой недостаточностью, либо полной неспособностью выработки особого фермента – глюкуронилтрансферазы (УДФГТ).
Причины
Первичной причиной заболевания является дефект гена, отвечающего за выработку фермента, в результате УДФГТ в организме отсутствует или находится в незначительном количестве. Заболевание врожденное, обнаруживается у обоих полов в равных соотношениях.
Классификация
Различают два типа заболевания:
- Первый тип. Полное отсутствие активности фермента УДФГТ. Для этого типа характерны мутационные изменения в гене, приводящие к образованию неполноценного фермента, который не способен выполнять свои функции. Впоследствии патологический УДФГТ активно разрушается. Все это приводит к накоплению токсичного билирубина и тяжелым последствиям для всего организма. Билирубиновая энцефалопатия 1-го типа подразделяется на четыре фазы.
- Второй тип (синдром Ариаса). Проявляется резким снижением активности УДФГТ при сохранении его нормального строения.
Симптомы
Первый тип. С первых часов жизни младенца проявляется тяжелой формой желтухи, не проходящей со временем и даже при интенсивном лечении. Быстро присоединяется билирубиновая энцефалопатия (токсическое поражение центральной нервной системы), приводящая к инвалидности ребенка и летальному исходу в первые два года жизни.
Клинические признаки синдрома Криглера-Найяра:
- Желтизна кожи и склер (ядерная желтуха).
- Судороги.
- Атетоз (непроизвольные движения конечностей, лица, головы).
- Нистагм (непроизвольные ритмичные движения глаз).
- Снижение умственной деятельности и замедление ее развития.
Первая фаза – сонливость у новорожденного, снижение сосательного рефлекса, замедление дыхания, постепенно присоединяются длительные временные промежутки его остановки. Кожа синюшного оттенка, возможны рвота, срыгивания, длительный монотонный крик.
Вторая фаза – появление симптомов поражения головного мозга. Наблюдается ригидность мышц затылка, статичность, характерно положение тела с «не сгибающимися» конечностями, ручки сжаты в кулачки, подергиваются лицевые мышцы. Отмечается исчезновение сосательного рефлекса, набухание большого родничка. Ребенок сильно кричит и не реагирует на звуки. Возможны судороги, апноэ, летаргия.
Третья фаза – симптомы спастики мышц исчезают полностью или частично – мнимое улучшение состояния.
Четвертая фаза – отмечается ухудшение состояния. Нарастает клиническая картина выраженных нарушений нервной системы: паралич, апноэ, парезы, рассеянность внимания. Младенец не реагирует на звуки и другие раздражители, не фокусирует взгляд.
Без своевременного лечения младенцы погибают, не дожив до года.
Второй тип проявляется более сдержанной симптоматикой. Жалобы в первые года жизни могут отсутствовать, а отмечаться только в подростковом возрасте. Характерна желтуха без нарушений со стороны нервной системы и других органов. Редки случаи возникновения энцефалопатии после стресса, либо интеркуррентных инфекций.
Течение болезни
Первый тип синдрома Криглера-Найяра отличается злокачественным прогрессированием заболевания, быстро приводящего к гибели ребенка.
Второй – более благоприятен, болезнь прогрессирует крайне медленно или остается стабильной в течение всей жизни.
Диагностика:
Диагностические мероприятия включают:
- Осмотр с физикальным исследованием.
- Лабораторная диагностика крови с определением уровня билирубина различными методиками.
- Дуоденальное зондирование.
- Хроматографическое исследование желчи.
- Холеграфия (пероральная).
- Биопсия печени.
- Проведение ДНК-диагностики с целью выявления мутаций гена UGTIAI.
Лечение
Терапия направлена на снижение уровня токсичного билирубина, повышение его выделения из организма. Проводится комплексное лечение, включающее лекарства, стимулирующие активность глюкуронилтрансферазы, переливания крови, вливание плазмы, фототерапию, пересадку печени.
К комплексу лечения второго типа добавляют симптоматические препараты, снижающие неврологические проявления: фенобарбитал, клофибрат. Назначают процедуры плазмофереза.
Прогноз
Пациенты с первым типом болезни редко живут больше двух лет. Смерть наступает от последствий билирубиновой энцефалопатии. Редко больные дети доживают до подросткового возраста.
Прогноз благоприятнее и продолжительность жизни выше у детей со вторым типом синдрома. Она зависит от степени тяжести болезни. Энцефалопатия возникает редко, неврологические нарушения отсутствуют, качество жизни значительно не нарушается.