Заболевание легче поддается лечению в случае ранней диагностики, поэтому важно знать: что это такое, как протекает у детей, симптомы, варианты диагностики и лечения патологии.
Что такое гепатолентикулярная дегенерация?
Это редкое заболевание, передающееся по наследству, характеризуется избыточным накоплением и отложением меди в тканях (преимущественно – в печени и структурах нервной системы).
Гепатолентикулярная дегенерация встречается довольно редко (средняя распространенность – 1 на 25000 населения). В основе болезни – нарушение процесса выведения меди из организма, в результате чего со временем развивается хроническая интоксикация этим металлом.
Медь откладывается во многих органах, приводя к изменению клеток. Ее накопление в клетках печени ведет к развитию воспалительного процесса, к некрозу, затем нормальная ткань органа замещается на плотную фиброзную.
Поражение нервной системы заканчивается гибелью нейронов с формированием в веществе головного мозга полостей. Печень и ЦНС поражаются чаще всего и по выраженности клинических проявлений с их стороны можно определить дальнейший прогноз.
При разрушении некротизированных клеток происходит выделение большого количества меди, что приводит к ее избытку в кровотоке, а в последующем – к гемолизу эритроцитов и недостаточности функций печени.
Симптомы у взрослых
В зависимости от проявлений заболевание можно разделить на три формы: с поражением печени, нервной системы и смешанный вариант.
У болезни Вильсона-Коновалова имеются две стадии:
- латентная (когда визуально признаки болезни не обнаруживаются, но по результатам лабораторных исследований можно поставить диагноз);
- стадия клинических проявлений (у больного при осмотре можно увидеть характерные симптомы заболевания).
Дебют клинических проявлений заболевания начинается с печеночной симптоматики (это прослеживается у половины больных). Далее медь начинает накапливаться в центральной нервной системе, что сопровождается признаками нарушения психических и неврологических актов.
Начало заболевания с симптомов нарушения ЦНС наблюдается в среднем у 35% больных. Это можно объяснить тем, что накопление меди в печени может длительно протекать бессимптомно, в какой-то степени это зависит от уровня компенсаторных возможностей органа.
У оставшейся части пациентов патология начинается с гемолиза эритроцитов, что лабораторно подтверждается анемией, затем наблюдаются и другие нарушения со стороны крови.
В роговице глаза медь при болезни Вильсона-Коновалова начинает накапливаться в одно время с появлением нарушений нервно-психической деятельности.
Отложение меди в печени может проявляться любыми патологиями органа.
Их выраженность у пациента варьирует от бессимптомного течения (когда только по анализам можно обнаружить небольшие отклонения показателей от нормы) до тяжелейших клинических проявлений цирроза, гепатита с осложнениями. Признак поражения печени могут быть у пациента с болезнью Вильсона-Коновалова задолго до симптомов нарушения работы нервной системы.
Гепатоцеребральной дистрофии свойственны разнообразные симптомы со стороны работы нервной деятельности. Они могут нарастать длительно и практически не беспокоить пациента, в тяжелых случаях за несколько месяцев накопление меди в ЦНС приводит к инвалидизации больного. При осмотре выявляются различные нарушения поведения, психики и неврологические расстройства.
В клинике болезни Вильсона-Коновалова чаще всего наблюдается мозжечковая атаксия с тремором и другими характерными симптомами для поражения мозжечка, явления паркинсонизма. Пик выявления неврологических нарушений у больных – с 10 до 35 лет.
Как развивается заболевание у детей?
При осмотре нарушения выявляются в возрасте старше 5 лет, но по лабораторным показателям уже с рождения можно наблюдать повышенные показатели печеночных трансаминаз.
Роль генетики и тип наследования
Болезнь Вильсона-Коновалова развивается вследствие наследственной мутации гена АТР7В, который располагается на 13 хромосоме. Этот ген отвечает за синтез медьтранспортирующей АТФ-азы определенного типа.
На сегодняшний день ученые выявили уже более 500 различных мутаций гена, большая часть из них имеет доказанную роль в развитии патологического процесса. Тип наследования болезни – аутосомно-рецессивный.
Признаки болезни Вильсона-Коновалова наиболее яркие у гомозиготных больных, у которых выявлено наличие двух одинаковых мутантных гена. При этом большая часть пациентов имеют разные мутации в хромосомах, каждая из которых передалась от одного из родителей.
Те лица, у которых от одного родителя достался ген с мутацией, а от второго – нормальный ген, не имеют никаких признаков заболевания и являются здоровыми, но при этом они носители мутантного гена. При рождении ребенка от такого же гетерозиготного носителя вероятность того, что он будет болен, составляет 25%. Половине от всех рожденных детей передаются мутантные гены.
Код МКБ 10
Болезнь Вильсона-Коновалова значится в МКБ 10 пересмотра под кодом Е83.0.
Диагностика гепатоцеребральной дистрофии
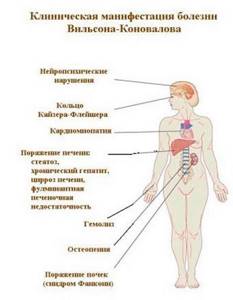
Одним из наиболее ярких клинических признаков гепатоцеребральной дистрофии являются симметричные изменения радужной оболочки глаза: появляется медный ободок – кольцо Кайзера-Флейшера.
Отмечается практически у всех больных на момент проявления неврологической симптоматики, при печеночных признаках болезни Вильсона-Коновалова кольца на радужке выявляются у половины пациентов.
В детском возрасте эти изменения могут принимать не форму кольца, а иметь вид мелких вкраплений на радужке или располагаться в виде дуг.
Для диагностики патологии обмена меди используются в основном лабораторные методы, второстепенный характер имеют инструментальные исследования. Основными являются:
- Общее и свободное количество меди в сыворотке крови.
- Концентрация меди в печеночных клетках.
- Суточное выделение меди из организма с мочой.
- Церулоплазмин сыворотки крови.
- Содержание меди в цереброспинальной жидкости – используется нечасто, но полученные значения могут дать сведения об уровне поражения головного мозга отложениями металла.
- ДНК-диагностика (генетика).
Инструментальные данные используются уже на этапе клинических проявлений болезни Вильсона-Коновалова, позволяют получить специалисту информацию о состоянии органа. Используются:
- МРТ и КТ – для оценки изменений со стороны центральной нервной системы. При оценке трехмерного изображения выявляются характерные очаги, зоны диффузной атрофии коры, изменения вещества ствола мозга.
- Биопсия печени. Манипуляция инвазивна и опасна осложнениями, поэтому ее производят только в случае, если имеются затруднения в постановке окончательного диагноза, а все другие неинвазивные исследования уже были пройдены пациентом. Также гистологическое исследование печени рекомендуется пациентам для диагностики других болезней печени, не связанных с основной обменной патологией.

Клинические рекомендации по лечению
В терапии гепатолентикулярной дегенерации используются препараты, которые разными путями ускоряют выведение меди из организма. Вместе с этим при болезни Вильсона-Коновалова обязательно назначается диета, которая направлена на употребление продуктов с низким содержанием меди. При тяжелом течении заболевания пациентам рекомендуется пересадка печени.
На этапе, когда клинические признаки болезни отсутствуют, целью назначенного лечения является не допустить их появления. Критерием эффективности является нормализация лабораторных показателей крови и мочи у пациента.
Если у больного имеются симптомы патологии печени или ЦНС, то главной задачей лечения будет предупреждение дальнейшего развития болезни Вильсона-Коновалова и регресс имеющихся проявлений.
Контроль эффективности проведенного лечения также осуществляется по лабораторным данным.
Все лечение делится на два этапа: начальная (стартовая) и поддерживающая терапия. Стартовая доза назначается для уменьшения выраженности симптоматики и нормализации анализов. При достижении нужных показателей при двукратном исследовании с разницей в три месяца можно переходить на поддерживающую терапию.
Нормальные показатели меди после проведенного лечения говорят о ликвидации накопленной меди из организма (кроме нервной системы). Об этом может говорить снижение до физиологического уровня трансаминаз печени – клетки органа перестают разрушаться и постепенно восстанавливаются.
Хелатная терапия
При базовом лечении используется d-пеницилламин, триентин. Помимо этих средств, применяются тетратиомолибдат и унитиол.
При лечении этими средствами нужно обязательно периодически проводить контроль уровня показателей общего анализа крови и мочи. При старте лечения это рекомендуется проводить раз в три дня, затем раз в неделю в течение одного месяца.
При резких отклонениях от нормы нужно снизить дозировку или подобрать другой препарат.
После приема лекарства в течение месяца контрольные анализы нужно сдавать раз в две недели в течение полугода, а затем достаточно будет ежемесячного обследования.
Также ведется наблюдение за биохимией крови – это необходимо как для контроля эффективности лечения, так и для оценки токсичности назначенного препарата для организма пациента.
Раз в год обязательно производится ультразвуковое исследование мочевыводящей системы.
Препараты цинка
Часто хелатные препараты используют в комбинации с соединениями цинка, так как это позволяет уменьшить концентрацию первых и снизить вероятность развития побочных эффектов. В РФ для лечения болезни Вильсона-Коновалова применяется препарат Цинктерал.
Экстракорпоральная гемокоррекция
Назначается при выраженных нарушениях функций печени: недостаточность органа, тяжелая форма гепатита или декомпенсированная форма цирроза, массивный гемолиз эритроцитов, тяжелые нарушения со стороны внутренних органов или нервной системы. Применяется в качестве экстренной терапии для быстрого снижения уровня свободно циркулирующей меди в крови и стабилизации состояния пациента.
Трансплантация печени
Показана пациентам с осложнениями и тяжелым течением болезни Вильсона-Коновалова, у которых нет положительного эффекта от назначенной терапии в течение 3 месяцев. В донорском органе нет никаких нарушений в метаболизме меди, поэтому такое радикальное оперативное вмешательство имеет хороший результат в виде регресса имеющейся симптоматики путем восстановлений нормального обмена меди.
После проведенного хирургического лечения выживаемость больных составляет примерно 80-86% в течение пяти лет. Пересадку органа можно произвести, взяв часть печени от живого донора, а также от умершего человека при условии гистологической совместимости.
Трансплантация печени показывает отличные результаты в том случае, если на первый план выходят печеночные симптомы. Неопределенность имеется при пересадке печени больным с преобладанием неврологических нарушений, так как у части больных тяжесть симптомов после хирургической операции снижается, а у другой группы больных остается на прежнем уровне.
Регистрируются случаи, когда после трансплантации неврологические нарушения нарастают, в особо тяжелых случаях приводят к инвалидизации пациентов. Важно следить у таких больных за получением иммуносупрессивной терапии, так как психические нарушения могут повлечь за собой отказ от таблеток и реакцию отторжения трансплантата.
Нужно ли соблюдать диету?
Медь находится практически во всех продуктах питания, но ее количества в них различны. Полностью исключить поступление этого соединения в организм нельзя, но ограничить вполне возможно. Специалисты рекомендуют исключить из рациона следующие продукты, в которых концентрация меди превышает максимально допустимое значение – 0,5мг на 100 грамм:
- орехи;
- гречневая крупа;
- шоколад;
- печень;
- шпинат;
- кальмары;
- ракообразные и моллюски;
- грибы.
 Орехи следует исключить из рациона
Орехи следует исключить из рациона
Внимательно нужно изучать составы поливитаминных и минеральных комплексов, различных пищевых добавок и БАДов – избегать те, что содержат медь в своем составе. Для пациентов с болезнью Вильсона-Коновалова в них находится токсическая дозировка металла.
Есть данные о том, что у больных, соблюдающих вегетарианскую диету, отмечается более позднее начало появления симптомов болезни Вильсона-Коновалова и они менее выражены, нет тенденции к быстрому прогрессированию.
Контролировать содержание металла нужно и в воде, особенно, если водоснабжение не централизованное. При повышенных значениях меди обязательно нужно использовать водоочистные приспособления. Для приготовления и хранения пищевых продуктов не надо использовать посуду из меди.
Продолжительность жизни при лечении
В случае своевременного лечения прогноз благоприятный: удается в большей части случаев добиться регресса основной симптоматики и нормализации лабораторных показателей пациента. Это достигается путем приема назначенных лекарств и строгого соблюдения диеты. Больные не отличаются от здоровых людей.
Прогноз при отсутствии терапии
На фоне отсутствия терапии заболевание прогрессирует, нарастают симптомы поражения внутренних органов, нервной системы. Резко сокращается продолжительность жизни при отсутствии лечения у больных ввиду тяжелых нарушений функций печени и ЦНС.
Основной причиной летального исхода у таких больных является фульминантная форма печеночной недостаточности, декомпенсация цирроза печени. В более редких случаях смерть пациента обусловлена тяжелыми неврологическими нарушениями.
Выводы
- Болезнь Вильсона-Коновалова имеет характерные симптомы, по которым можно распознать болезнь и начать терапию.
- При строгом соблюдении всех врачебных рекомендаций прогноз для выздоровления и жизни пациента благоприятный.
- Необходимо обследовать всю семью, если был зафиксирован случай болезни Вильсона-Коновалова: на досимптомной стадии патология легче поддается лечению и на фоне поддерживающей терапии можно добиться полного выздоровления.
Болезнь Вильсона-Коновалова: симптомы, диагностика, лечение и причины
Болезнь Вильсона-Коновалова является наследственной патологией обмена меди, наследуемой по аутосомно-рецессивному типу. Женщины и мужчины болеют приблизительно одинаково.
Информация для врачей. По МКБ 10 болезнь Вильсона-Коновалова шифруется под кодом E83.0. Обязательно указывается форма заболевания, выраженность неврологических проявлений, уровня печеночной недостаточности.
Причины
Причиной заболевания служит наследственный дефект генов, отвечающих за выработку церулоплазмина, участвующего в транспорте меди.
Вследствие недостатка белка-переносчика происходит избыточное отложение меди в подкорковых узлах головного мозга (прежде всего, в чечевицеобразном), печени.
Именно поэтому болезнь Вильсона-Коновалова носит другое название – гепатолентикулярная (гепатоцеребральная) дегенерация.
Симптомы
Заболевание, как правило, начинает развиваться в подростковом или юношеском возрасте.
На первый план выходят такие симптомы, как нарастающая мышечная ригидность, гиперкинезы различной формы и амплитуды, дрожание конечностей, туловища, головы, нарушениями речи и психики.
Иногда дополнительно имеют место эпилептиформные приступы. При общем осмотре нередко можно обнаружить болезненность и увеличение печени.
- Брюшная. Отсутствуют или слабо выражены неврологические симптомы. Имеют место лишь слабо выраженные гиперкинезы, изменения психики. На первый план выступает печеночная недостаточность. Заболевания быстро прогрессирует и практически всегда приводит к летальному исходу. При течение заболевания свыше 3 лет могут отмечаться неврологические проявления.
- Ригидно-аритмогиперкинетическая. Развивается чаще в детском возрасте. Представлена развитием мышечных контрактур, нарушениями речи, насильственным смехом, плачем, бульбарными нарушениями, задержкой психиеского развития. Прогрессирует стремительно, приводит к летальному исходу.
- Дрожательная. Развивается, как правило, у взрослых. Представлена преимущественно дрожью в руках, изменениями психики, гипотонией мышц. Протекает медленно, нередко имеют место эпилептиформные приступы. Средняя продолжительность жизни может составлять 15 лет и более.
- Дрожательно-ригидная. Самая частая форма болезни Вильсона-Коновалова. Протекает медленно, начала в юношеском возрасте. Отмечается нарастающая мышечная ригидность, гиперкинезы, ритмичное дрожание. Во сне симптомы, как правило. полностью проходят. Среднее течение заболевания 6-8 лет.
- Экстрапирамидно-корковая. Встречается крайне редко.
Диагностика
Диагностика гепатолентикулярной дегенерации основывается на обнаружении: изменения активности церулоплазмина плазмы крови, повышением суточной экскреции меди с мочой, наличие патогномичного признака – кольца Кайзера-Флейшера или его элементов, также нередко обнаруживается гипераминацидурия.
Также снижается уровень меди крови, однако одного этого признака недостаточно для постановки диагноза. Также зачастую можно провести семейный анализ и выявить факт наследования. Дифференциальный диагноз необходимо проводить с эпидемическим энцефалитом, ревматической хореей, торсионной дистонии, последствиями приемов некоторых лекарственных препаратов (прежде всего, нейролептиков).
Дифференциальный диагноз необходимо проводить с эпидемическим энцефалитом, ревматической хореей, торсионной дистонии, последствиями приемов некоторых лекарственных препаратов (прежде всего, нейролептиков).
Лечение
Лечение болезни Вильсона-Коновалова следует проводить препаратами, увеличивающими экскрецию меди. Среди них наибольшее применение нашли такие препараты как унитиол, пеницилламин. Также дополнительно проводится симптоматическая терапия миорелаксантами (для снижения мышечного тонуса), противоэпилептическими препаратами при необходимости, а также гепатопротекторными средствами.
Во всех случаях необходимо назначение также специальной диеты с ограничением меди. В обязательном порядке из рациона должны быть исключены огурцы, сухофрукты, орехи, продукты из печени животных и птиц. Ограничение накладываются на употреблении шоколада, некоторых морепродуктов.
На фоне лечения отмечается порой полное исчезновение симптомов заболевания. Своевременно начатое лечение позволяет увеличить продолжительность жизни больных.
Относительное значение имеет медико-генетическое консультирование при наличии данных анамнеза на наличие болезни Вильсона-Коновалова у ближайших родственников. Определяют концентрацию церулоплазмина в плазме крови и наличие дефектов генов, расположенной в 13-й хромосоме. При выявлении дефектных генов у обоих родителей можно предположить 25% вероятность рождения больного ребенка.
Синдром Вильсона-Коновалова: что это такое, симптомы и признаки, диагностика, лечение
Гепатолентикулярная дегенерация (другое название синдром Вильсона-Коновалова) – это редкое и сложное заболевание, которое развивается вследствие тяжелых метаболических расстройств в организме. Имеет наследственную природу. Встречается у одного человека на 100 тысяч.
Впервые недуг описал Сэмюель Вильсон в далеком 1912 году. В России большой вклад в изучение патологического процесса внес невролог Коновалов. Он создал классификацию заболевания. Ее используют и сейчас при формулировке клинического диагноза.
Чаще всего диагностируют в возрасте 5-45 лет. Хотя встречаются клинические случаи, когда патология проявлялась у маленьких детей либо пожилых людей. Рассмотрим патогенез, симптомы, формы и способы лекарственной терапии.
Этиология синдрома Вильсона-Коновалова
Заболевание носит генетическую природу. Синдром Вильсона-Коновалова и синдром Вильсона-Микити – это разные недуги. В последнем случае речь идет о патологии легких, которую диагностируют только у новорожденных детей. Проявляется развитием поздней кислородозависимости.
Причина формирования патологии кроется в генетике. Ген, который несет ответственность за возникновение недуга, располагается в 13-й хромосоме. Он принимает участие в перемещении меди. Заболевание Вильсона наследуется от родителей как рецессивный аутосомный признак, развивается вследствие даже незначительной мутации гена.
Особенность недуга в том, что заболеет ребенок только в том случае, если мама и папа являются носителями гена. Люди, имеющие в анамнезе один пораженный ген, не страдают от симптоматики патологии, но у них могут быть незначительные нарушения метаболизма меди.
В организме взрослого человека средняя концентрация меди около 100 мг. При этом потребность в сутки для нормального функционирования всех органов и систем 1-2 мг. На фоне чрезмерного поступления вещества в организм излишки всасываются печенью, вместе с желчью выводятся естественным путем.
При болезни Вильсона нарушаются сразу два процесса – это биологическая выработка белков, которые связывают желчь, и выведение естественным способом. Вследствие этого содержание вещества возрастает, медь откладывается в органах:
- Роговицы глаз.
- Почки.
- Печень.
- Головной мозг.
При критическом возрастании концентрации происходит токсическое поражение внутренних органов человека. Развивается цирроз печени, чаще всего крупноузловой формы. В головном мозге проявляется нарушение функциональности мозжечка, а в глаза формируется кольцо Кайзера-Флейшера/
Почему появляется болезнь?
Единственная причина формирования недуга – мутация определенного гена, который несет ответственность за метаболизм меди. В данном носителе ученые обнаружили более 100 возможных отклонений, поэтому анализ вероятных нарушений ДНК – неэффективный способ.
Предупредить недуг невозможно, профилактики не существует. Если ген имеется у обоих родителей, то у ребенка уже к 2-3 годам будут проблемы с нарушением работы печени.
Симптоматика
Гепатолентикулярная дегенерация имеет код по МКБ Е83.0. При клиническом описании патологического процесса медицинские специалисты обязательно указывают вид недуга, выраженность нарушений со стороны центральной нервной системы, печени.
Клиника развивается в раннем возрасте. Симптомы похожие на различные болезни железы. Чаще всего маленькие пациенты страдают от выраженного пожелтения кожных покровов, астении, анорексии. У девочек часто повышается температура тела. Сбить лекарственными средствами не получается.
Печень больного насыщается медью. После вещество начинает скапливаться в других органах, что приводит к сбою в работе ССС, ЖКТ, ЦНС. В последнем случае скопление меди в нервной системе негативно влияет на моторику, мимику, координацию движений.
Интеллект у ребенка полностью сохраняется не на фоне всех форм. Еще болезнь влияет на поведение – проявляются агрессивность и раздражительность.
Когда в глазах увеличивается концентрация меди до критического уровня, то происходит формирование кольца коричневого цвета. Его можно диагностировать посредством щелевой лампы, но только у детей старше 5-летнего возраста.
Гепатолентикулярная дегенерация сопровождается клиническим полиморфизмом, в процесс вовлекаются органы выделительной системы. У патологии присутствуют рецессивные признаки, которым предшествуют расстройства ЖКТ.
Виды заболевания и их симптомы
В зависимости от клинических проявлений в медицинской практике выделяют три формы патологии Вильсона-Коновалова – болезнь, которая нарушает работу печени, заболевание, поражающее ЦНС, смешанный вид. Согласно типам у больного преобладает та либо иная клиника.
Печеночный вид
Печеночный тип иногда называют брюшным. Он развивается у людей до 40-летнего возраста, сопровождается расстройством работы печени. Симптоматика схожа с клиническими признаками циррозного поражения.
В 80% клинических картин печеночный тип у пациентов протекает с проявлением таких симптомов:
- Слабость, постоянная усталость.
- Повышенное газообразование.
- Боль в области печени тупого характера.
- Увеличение объема жидкости в брюшной полости.
- Из носа периодически идет кровь.
- Пожелтение кожного покрова.
- Лихорадочное состояние.
- Увеличивается в размере селезенка.
- Утолщаются пальцы верхних/нижних конечностей.
В остальных 20% клинических картин к этим симптомам могут добавляться другие признаки заболевания.
Неврологический тип
Данный вид патологического процесса отличается другими проявлениями. Они начинают развиваться в раннем возрасте. У детей наблюдается мышечная ригидность, расстройство речи, постепенно снижаются интеллектуальные способности. На фоне неврологического типа бывают периоды обострения и затишья.
При обострении патологии в 10-25 лет у больных проявляется тремор конечностей, нарушение темпа речи из-за затруднения издавать расчлененные звуки. Пациенты медленно пишут, читают, разговаривают, часто бесцельно двигают руками.
Редкие клинические проявления
Любой вид синдрома Вильсона-Коновалова может дополниться другой симптоматикой. Она выявляется в 20% случаев. Симптомы – увеличение грудных желез у мужчин, нарушение слухового восприятия, чрезмерная хрупкость костей, что приводит к постоянным травмам, посинение ногтевых пластин, кожных покровов. Также нарушается работа почек, проявляется гемолитическая форма анемии.
Течение патологии
В соответствии с общепринятой классификацией заболевание бывает острого и хронического течения. Врачи также выделяют еще скрытую стадию, длительность которой не более 7-летнего периода. На фоне латентной стадии симптоматика имеется, однако она не оказывает существенного влияния на качество жизни больного.
В некоторых случаях патология никак не проявляет себя до 5-летнего возраста ребенка. Пик заболевания приходится на возраст 8-15 лет, однако уже с рождения имеются нарушения работы печени.
В таблице представлены особенности заболевания в зависимости от типа течения:
| Тип течения | Описание |
| Острый | Заболевание ярко проявляется уже в раннем возрасте, симптомы острые, выражены сильно, возникают комплексно и сразу. Организм пациента быстро угасает, медикаментозная терапия не приносит облегчения. Риск летального исхода при такой клинике свыше 90%. Несмотря на развитость медицинской науки, способов помочь таким пациентам еще не придумали. |
| Хронический | Отличается этот вид медленным прогрессированием. Вначале может быть скрытая стадия. Сначала страдает печень, после ЦНС. В подростковом возрасте имеются проблемы с координацией движения. Выраженность симптоматики средняя, прогрессирует клиника медленно. |
Методы диагностики
Диагностикой патологии занимаются несколько врачей – гепатолог, нефролог и гастроэнтеролог. Так как болезнь связана с наследственным фактором, то в диагностический процесс привлекают генетиков. Также требуются профессиональные консультации дерматолога, офтальмолога и ряда других специалистов, чтобы выявить осложнения.
Для начала собирают анамнез больного, осуществляют физикальный осмотр. На основании результатов дают клинические рекомендации, назначают соответствующие методы диагностики.
Диагностика включает в себя исследование урины и крови (определяют уровень меди). У офтальмолога с помощью щелевой лампы определяют отсутствие/наличие кольца Кайзера-Флейшера. Дополнительно проводится УЗИ, МРТ головного мозга, забор биоптата из печени.
Способы лечения
Терапия ориентирована на уменьшение поступления вещества в организм пациента. Для этого рекомендуется исключить из рациона продукты, которые содержат медь. Из меню убирают баранину, свинину, кальмары, креветки, крабов, грибы, сухофрукты, шоколад. Можно кушать молочные продукты, фрукты, овощи, курицу, куриные яйца, хлебобулочные изделия.
Чтобы снизить содержание меди в организме, когда оно неуклонно растет, медицинские специалисты рекомендуют прием медикаментов противовоспалительного, иммуносупрессивного свойства. В схему терапии входят лекарства желчегонного свойства, антиоксиданты, препараты с цинком.
Дозировка пациенту устанавливается индивидуально. Обычно назначают самую маленькую и отслеживают динамику улучшений. Дополнительно рекомендуется прием витаминно-минеральных комплексов. Перед покупкой оных нужно внимательно изучать инструкцию, в составе не должно быть меди.
Если медикаментозное лечение не дает нужного эффекта, самочувствие больного ухудшается, то единственный способ – это трансплантация печени. После хирургического вмешательства продолжают прием поддерживающих медикаментов.
Возможные осложнения
Лечение лучше начинать сразу после выявления заболевания, даже когда выраженные симптомы отсутствуют. Это позволит предупредить клинику, способствует замедлению прогрессирования недуга.
К наиболее распространенному осложнению относят патологии печени. Они проявляются симптомами:
- Пожелтение кожи.
- Деформация пальцев верхних и нижних конечностей.
- Расширение вен на передней брюшной стенке.
- Отечность ног.
Часто у больных выявляется кровотечение в ЖКТ. Развивается печеночная недостаточность, к симптоматике которой относят нарушение сна, расстройства поведения, в тяжелых случаях наблюдается печеночная кома.
К осложнениям относят нарушения неврологического характера. Это мышечная дистония, дизартрия, расстройства поведения, личности, эпилептические припадки. У женщин нарушается фертильность.
Прогноз и профилактические мероприятия
Благоприятный прогноз возможен только в одном случае – если заболевание обнаружили в раннем возрасте, сразу же начали терапию высокоэффективными медикаментами. Важно начать терапевтический курс до поражения ЦНС и печени.
Прием медикаментов улучшает функциональность печени, нивелирует негативную симптоматику со стороны ЦНС. Уже спустя 6 месяцев от начала терапии значительно улучшается самочувствие больного. Более выраженные улучшения видны через 2-3 года.
Если лечение отсутствует, начато поздно либо эффективность терапии недостаточна, то летальный исход наступает в 35-40-летнем возрасте. Как правило, причиной выступает печеночная недостаточность. При серьезных поражениях железы требуется трансплантация органа. Чем раньше ее сделать, тем лучше приживется орган. Выживаемость среди больных 20-ти лет составляет 80%.
Специфических профилактических мер нет, потому что болезнь связана с наследственностью человека. Тем, кто находится в группе риска, рекомендуется периодически посещать врача, проходить обследования, заниматься спортом, полностью отказаться от употребления алкогольной продукции.
Болезнь Вильсона
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях.
Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году.
Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг.
95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином.
Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.).
При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
- брюшная
- ригидно-аритмогиперкинетическая
- дрожательно-ригидная
- дрожательная
- экстрапирамидно-корковая
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте.
В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств.
С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди.
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов.
Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза).
Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца.
В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов).
В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия.
В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т.д.).
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.