Синдром Марфана — генетическое заболевание, которое характеризуется поражением соединительной ткани.
Встречается заболевание у детей с частотой 1:5000. Мальчики и девочки болеют одинаково.
В 75% случаях регистрируют семейный тип наследования. Риск заболевания возрастает, если возраст отца достигает 35 лет и выше.
Причины возникновения заболевания
Синдром Марфана — аутосомно-доминантное заболевание, то есть дети, унаследовавшие поражённый ген, будут страдать данным заболеванием. Дефектный ген FBN1 кодирует белок фибрилин-1. Такой белок, связываясь с другими белками, образует трансформирующий фактор роста бета. Накопление данного фактора негативно сказывается на функции органов и систем, вызывая симптомы заболевания.
Механизм развития заболевания
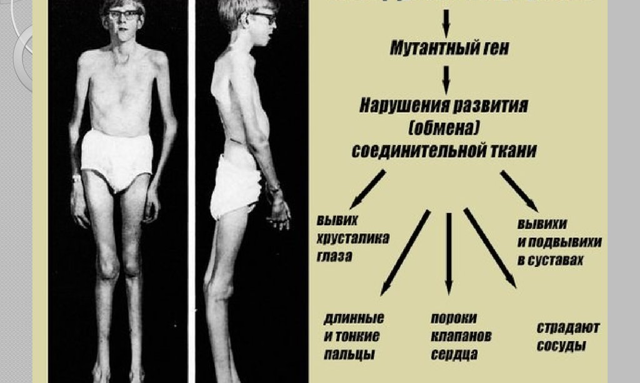
Заболевание развиваться из-за дефекта гена FBN1, который располагается в 15-й хромосоме. Данный ген кодирует белок фибрилин-1, который влияет на функционирование эластичных волокон, целостность соединительной ткани. Эластичные волокна находятся практически в каждом органе, но наибольшее их количество сконцентрировано в аорте, связках и в глазном аппарате глаза. Именно поэтому данные органы поражаются при заболевании в первую очередь. Снижение уровня белка фибролита-1 осуществляет трансформирующий фактор роста бета. Такой фактор накапливается в сердце, сосудах, лёгких, вызывая характерные симптомы генетического заболевания.
Какие различают формы?
Проявления синдрома Марфана встречаются во многих органах и системам. На основе полиморфизма клинической картины составлена классификация.
По форме:
- стёртая: выявляются симптомы в 1 — 2 системах;
- слабовыраженная: проявления в 3 — 4 системах;
- выраженная: отмечаются клинические симптомы в более чем 4 системах организма.
По степени тяжести:
- лёгкая;
- средней степени;
- тяжёлая.
Клиническая картина
Для данного заболевания характерен внешний вид больного. Мальчики при рождении имеют длину тела около 53 см, а во взрослом возрасте достигают 190 см. Девочки рождаются с длиной тела 52,5 и вырастают до 175 см.
Таким образом, больные данным генетическим заболеванием имеют высокий рост.
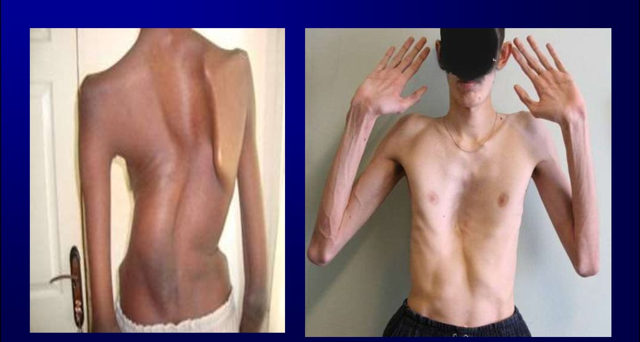
Помимо роста, у детей отмечают непропорционально длинные конечности и короткое тело, удлинённые паукообразные пальцы, вытянутое лицо и форма черепа, астеническое телосложение, снижение подкожно-жировой клетчатки.
Для синдрома Марфана характерно многообразие клинических симптомов.
Поражение костной системы
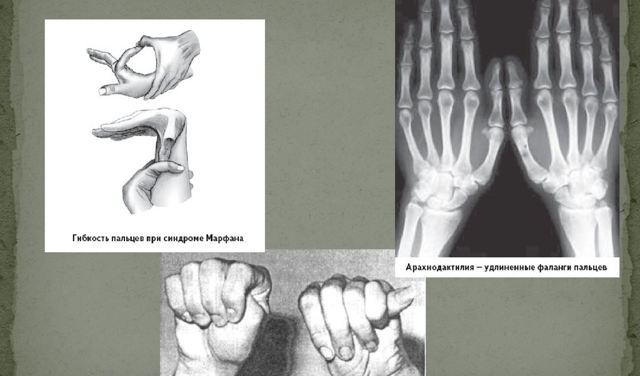
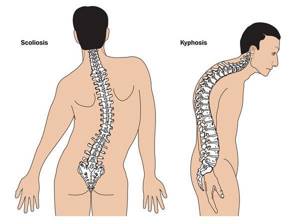
Наиболее часто поражается при заболевании именно костная система. В симптомокомплекс можно включить высокий рост, непропорциональное тело, арахнодактилия (паукообразные пальцы). У детей с данным синдромом развивается сколиоз (искривление позвоночника в сторону). Нередко выявляется деформация грудной клетки, изменения могут быть как внутрь (воронкообразная грудь), так и наружу (килевидная). Высокое нёбо, неправильный прикус у детей развивается, так как форма черепа имеет вытянутую форму. Такие изменения лицевого скелета способствуют нарушению речи. Плоскостопие также является симптомом болезни Марфана. У детей отмечается гипермобильность суставов (чрезмерная гибкость). Пациенты могут предъявлять жалобы на боль в суставах и костях.
Поражение глазного аппарата
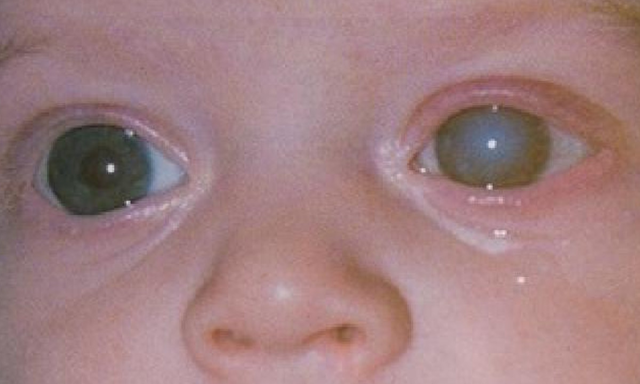 У 80% больных синдромом Марфана выявляют изменение положения хрусталика.
У 80% больных синдромом Марфана выявляют изменение положения хрусталика.
Наиболее часто у детей офтальмолог при помощи щелевой дампы выявляет подвывих хрусталика. Помимо дислокации хрусталика, выявляется астигматизм, близорукость, глаукома в раннем возрасте.
Поражение сердечно-сосудистой системы
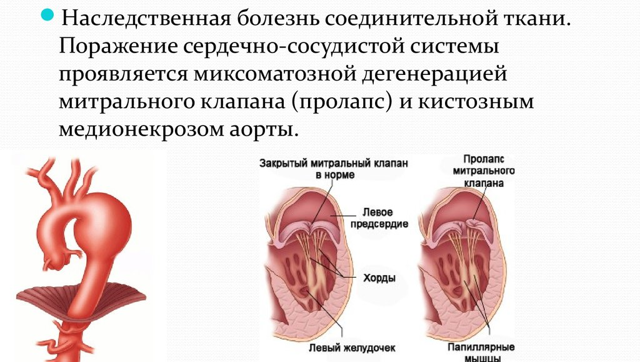
Самыми грозными являются изменения со стороны сердечно-сосудистой системы. Дети могут жаловаться на повышенную утомляемость, учащение сердцебиения и дыхания, нарушение ритма сердца, возможно появление болей в области сердца. Появление шумов при аускультации даёт повод для более тщательного обследование пациента. Наиболее опасным является изменение аорты: аневризмы (выпячивание стенки) или расслоение верхней её части. Но такие явления никак себя не проявляют. Особого внимания требуют беременные женщины, так как во время родов возможно расслоение аорты и смертельный исход. Поэтому необходимо каждые 6 недель проводить ультразвуковое исследование сердца и сосудов, для динамических замеров диаметра корня аорты.
Поражение лёгких
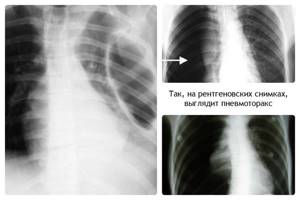
При синдроме Марфана возникает риск возникновение спонтанного пневмоторакса. Такое состояние характеризуется изменением давления в плевральной полости. Пациент чувствует боль в грудной клетке, наблюдается одышка, побледнение кожного покрова. Ребёнок не может глубоко вдохнуть. При отсутствии лечения пневмоторакс приводит к смерти больного.
Поражение нервной системы
При синдроме Марфана поражается твёрдая оболочка спинного мозга, а именно происходит её ослабление или растяжение (эктазия). Такое изменение несёт в себе целый ряд клинических проявлений: головная боль, боль в спине и конечностях.
На рентгенологическом исследовании такие изменения на ранних стадиях не видны, поэтому диагностический поиск может зайти в тупик.
Данное поражение может наблюдаться лишь при более эффективных методиках обследования пациента, таких как магнитно-резонансная и компьютерная томография.
Диагностика заболевания

- Консультация генетика, который собирает необходимый для диагностики семейный анамнез. Применяет специальные фенотипические тесты.
- Консультация ортопеда. Ортопедический осмотр выявляет сколиоз, плоскостопие, деформацию грудной клетки.
- Консультация кардиолога с проведением ЭХО-КГ (ультразвуковое исследование сердца). Данный метод позволяет визуализировать: пролапс (прогибание створок клапана) митрального или аортального клапана с явлением регургитации (обратный ток крови), расширенную аорту или её аневризма (выпячивание стенки).
- Магнитно-резонансная томография для выявления изменений со стороны твёрдой оболочки спинного мозга.
Для точной диагностики заболевания используются большие и малые критерии.
Диагноз может быть установлен при наличии 2 больших и 1 малого критерия в разных системах организма человека.
Большие критерии:
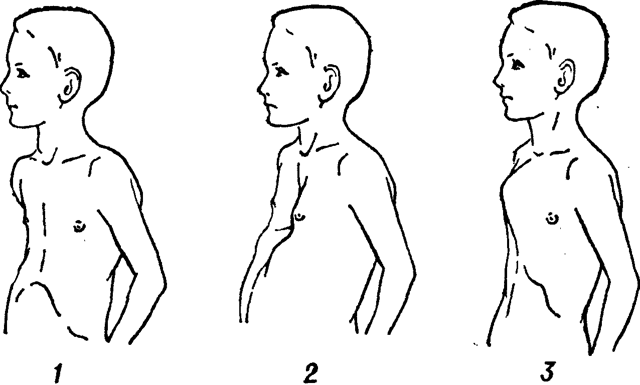
- деформация грудной клетки (килевидная или воронкообразная);
- преобладание нижней части тела над верхней;
- подвывих хрусталика;
- расширение диаметра восходящей части аорты;
- расслоение аорты;
- эктазия твёрдой мозговой оболочки;
- клинически диагностированное заболевание Марфана у родителей, сестры, брата ребёнка;
- мутация гена.
Малые критерии:
- сколиоз;
- плоскостопие;
- гипермобильность суставов;
- высокое нёбо;
- вытянутое лицо и череп;
- увеличение размера глазного яблока;
- недоразвитие роговицы;
- пролапс митрального клапана;
- расширение аорты или лёгочного ствола;
- спонтанный пневмоторакс;
- стрии, образующиеся без видимых причин.
Дифференциальная диагностика
Проводят дифференциальную диагностику с заболеваниями, имеющими схожую клиническую картину.
- Синдром Билса. Данная болезнь (врождённая контрактурная арахнодактилия), характеризуется наличием дефекта в гене FBN2, кодирующий белок фибрилин 2. Клинические проявления при таком состоянии практически не отличаются от синдрома Марфана. Единственным отличием служит отсутствие поражения зрения при синдроме Билса. И на первое место выходит изменение пальцев рук с развитием контрактур (невозможность движений).
- Гомоцистинурия — наследственное заболевание, в основе которого лежит нарушение обмена серосодержащих аминокислот. Диагноз можно поставить уже в раннем возрасте, опираясь на клинические проявления. Помимо схожих с синдромом Марфана симптомов, наблюдаются: отставание в физическом и нервно-психическом развитии, умственная отсталость, судороги, гиперкинезы (размашистые движения), поведенческие нарушения. Внешний вид очень схож с синдромом Марфана, но определяющая роль принадлежит умственной отсталости, которая наблюдается при гомоцистинурии.
- Синдром Элерса — Данлоса, который характеризуется гиперэластичностью кожи. Развивается при нарушении синтеза белка коллагена. Наряду со схожими симптомами синдрома Марфана, у пациента на первое место выходит поражение кожного покрова. Кожа имеет высокую эластичность и растяжимость, а также хрупкость и ранимость. Незначительные микротравмы и порезы длительно заживают и способны оставлять после себя рубцы.
- Синдром Лойса — Дитца. Такой синдром проявляется при дефекте в генах, отвечающих за синтез транформирующего фактора роста бета-1. Отличительной особенностью данного заболевания является гипертелоризм (широко посаженные глаза), порок развития лицевого скелета «волчья пасть» (расщепление твёрдого нёба).
О возможности лечения
Специфического лечения синдрома Марфана на сегодняшний день не существует. Но новые препараты и технологии помогают продлить жизнь пациента. Лечение при данном заболевании симптоматическое, то есть проводится при проявлении тех или иных симптомов.
Важнейшим компонентом лечения являются регулярные осмотры врачей, прежде всего, кардиолога для предотвращения развития аневризмы аорты и её расслоения.
Для реализации данной цели используются препараты, уменьшающие частоту сердечных сокращений (группа бета-блокаторов), а также лекарственные средства для снижения кровяного давления (ингибиторы ангиотензин-превращающего фермента).
Если наблюдается расслоение аорты или аневризма, то резко повышается риск разрыва аорты и летального исхода. В такой ситуации требуется хирургическая коррекция аорты. Благодаря таким операциям, существует вероятность продлить жизнь пациенту.
Нарушение деятельности скелетной мускулатуры и костной системы также поддаётся лечению. При болевом синдроме используют болеутоляющие средства и миорелаксанты. Данные изменения, характерные для синдрома Марфана, имеют хороший результат после физиотерапевтического лечения и курса массажа.
При развитии спонтанного пневмоторакса пациенту необходима срочная помощь и госпитализация в торакальное отделение хирургического стационара.
Для коррекции зрения рекомендуется ношение корректирующих очков. Если произошло развитие глаукомы, то необходимо оперативное лечение.
Профилактика
Специфической профилактики не существует. Но имеется возможность пренатальной генетической консультации для оценки риска рождения больного ребёнка.
Беременные женщины, страдающие синдромом Марфана, должны регулярно обследоваться не только в женской консультации, но и у врачей-специалистов с применением инструментальных методов диагностики.
Больные дети также пожизненно наблюдаются у докторов, для предотвращения развития осложнений. Физические нагрузки рекомендуют ограничивать.
Заключение
Прогноз и течение синдрома Марфана зависит от выраженности поражения гена и недостаточного синтеза белка фибрина.
До 90% больных синдромом Марфана не доживают до 50 лет.
При отсутствии должной и регулярной диагностики у пациента резко повышается риск внезапной смерти. Новые технологии в современной медицине могут существенно продлить жизнь человека и улучшить её качество.
Мы приложили много усилий, чтобы Вы смогли прочитать эту статью, и будем рады Вашему отзыву в виде оценки. Автору будет приятно видеть, что Вам был интересен этот материал. Спасибо!
(3
Синдром Марфана — симптомы и лечение
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровикова О. И., гинеколога-эндокринолога со стажем в 10 лет.
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани.[1]
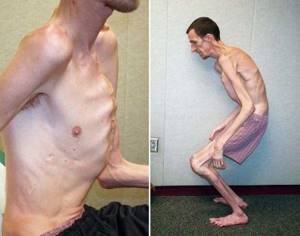
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1.[2]
- Наследование заболевания происходит по аутосомно-доминантному типу, характеризуется высокой пенетрантностью (частотой появления гена) и различной экспрессивностью.[5]
- Соотношение представителей мужского пола и женского одинаковое.
- Пример родословной с аутосомно-доминантным заболеванием[5]
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия).[1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения.[3]
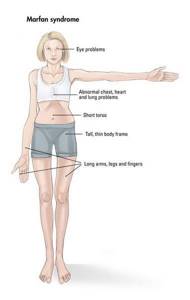
Кожа имеет повышенную растяжимость, разболтанные суставы.
У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы).[4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
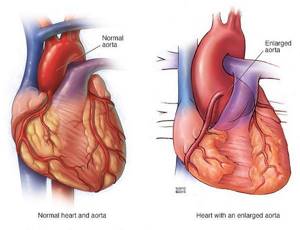
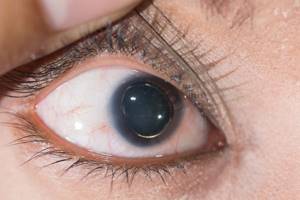
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте.[1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек.[2]
В четверти случаев зарегистрировано снижение интеллекта, у половины пациентов выявляются нарушения эмоционально-волевой сферы. Часто появляются депрессивные состояния, нейроциркуляторная дистония.[3]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации.[3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов.
Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани).
[4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта.[3]
- Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях.[3]
- Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией.[6]
- У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами.[4]
- Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы.[3]
- У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты).[7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности.[3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
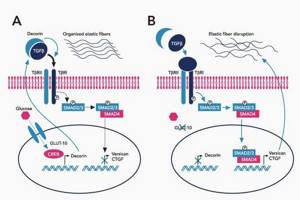
- Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
- В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
- При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни.[4]
- Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
- Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана.[6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция.[6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин.[7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана.
Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов.
[4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур.[3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны.[3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению.[3]
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
- Выделяют различные типы по степени тяжести:
- Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
- По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика.[7]
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана.[6]
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности).[7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
- Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1.[5]
- Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания.[1]
- Способы обнаружения арахнодактилии:[3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза.[3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз.[3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога.
В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани.
Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне.[3]
Оценка системных признаков вовлечённости соединительной ткани
| Совместное наблюдение положительных признаков Steinberg и Walker-Murdoch | 3 |
| Признак Steinberg и Walker-Murdoch отдельно друг от друга | По 1 |
| Килевидное искривление грудной клетки | 2 |
| Воронкоподобное искривление, либо асимметрия грудной клетки | По 1 |
| Медиальное смещение медиальной лодыжки, приводящее к уплощению стопы | 2 |
| Уплощение стопы | 1 |
| Спонтанный пневмо- и гидроторакс (скопление воздуха и жидкости в плевральной полости) | 2 |
| Расширение дурального мешка в крестцовом и поясничном отделах | 2 |
| Подтверждённая на рентгенограммах протрузия вертлужной впадины любой степени | 2 |
| Уменьшение отношения верхней и нижней частей туловища (< 1 у пациентов до 5 лет; < 0,95 в 6-7 лет; < 0,9 в 8-9 лет; < 0,85 у пациентов старше 10 лет) + отношение ширины размаха верхних конечностей к росту > 1,05 + искривление позвоночника I-II степени. | 1 |
| Сколиоз или кифосколиоз | 1 |
| Уменьшение выпрямления в локтевом суставе до 170 градусов и менее | 1 |
| Присутствие трёх черепно-лицевых дизморфий из пяти (долихоцефалическая форма черепа, впалые глаза, антимонголоидный разрез глазных щелей или смещение глазных щелей вниз, уменьшение размеров скуловых костей, ретрогнатия) | 1 |
| Растяжки на коже | 1 |
| Миопическая патология более трёх дптр | 1 |
| Пролапс (прогибание створок) митрального клапана | 1 |
Марфана синдром
Синдром Марфана (СМ), MARFAN SYNDROME (MFS) — генетическое заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы, скелета и глаз. Тип наследования аутосомно-доминантный. Распространенность в популяции около 2-3 случаев на 10 000. Синдром диагностируется во всем мире, в любых этнических группах.
Антуан Марфан, описавший этот синдром у пятилетней девочки, описывал его как долихоморфию, то есть удлинение конечностей по отношению к туловищу, однако позже Виктор Мак-Кьюсик показал, что это болезнь всей соединительной ткани. Диагноз ставится на основании критериев, принятых в 1996 г.
Предлагается различать особо важные для диагностики симптомы (их называют большими критериями) и менее важные симптомы, присутствие которых считают лишь вовлечением той или иной системы органов в болезнь. Если семейный анамнез не отягощен, то для постановки диагноза требуется наличие больших критериев по двум системам органов и вовлечение третьей.
Если в семье уже были случаи подобного заболевания, то для постановки диагноза достаточно одного генетического критерия и одного большого критерия в какой-нибудь системе органов с вовлечением другой системы. Поражение скелета считают большим критерием, когда имеются хотя бы четыре из восьми типичных проявлений.
Для других систем органов достаточно одного характерного поражения, чтобы оно считалось большим критерием.
Рост. Как правило, больные высокорослые, с длинными тонкими конечностями, слабо развитой подкожной клетчаткой и мышечной гипотонией. У мальчиков средняя длина тела при рождении—53 см, окончательный рост — 191 см; у девочек — 52,5 см и 175 см соответственно.
Скелет.
Составляющие большого критерия:
- килевидная или воронкообразная грудная клетка,
- требующая оперативного лечения;
- преобладание длины нижней части тела над длиной верхней или отношение размаха рук к росту выше 1,05;
Малые критерии:
- симптом запястья (способность обхватить запястье большим пальцем и мизинцем так, чтобы их дистальные фаланги перекрывались) и симптом большого пальца (кончик сжатого в кулаке большого пальца выглядывает наружу);
- сколиоз более 20 или спондилолистез;
- неполное разгибание в локтевом суставе (менее 170 );
- плоскостопие;
- протрузия вертлужной впадины.
- умеренно выраженная воронкообразная грудная клетка;
- разболтанность суставов;
- узкое высокое небо и скученность зубов;
- характерное «птичье» лицо (вытянутое и узкое за счет долихоцефалии и недоразвития скуловых костей, с энофтальмом и антимонголоидным разрезом глаз).
- О вовлечении скелета говорят, если у больного имеются хотя бы два проявления, составляющих большой критерий, либо одно проявление, относящееся к большому критерию, и два малых критерия.
- Глаза.
- Большой критерий:
- подвывих хрусталика, как правило, со смещением вверх и c дефектом цинновой связки.
Малые критерии:
- уплощение роговицы;
- увеличение переднезаднего размера глазного яблока;
- гипоплазия радужки или ресничной мышцы с ослаблением миоза.
- О вовлечении глаз говорят при наличии хотя бы двух малых критериев.
- Сердечно сосудистая система.
- Большие критерии:
- расширение восходящей аорты (с аортальной недостаточностью или без нее);
- расслаивающая аневризма восходящей аорты.
Малые критерии:
- пролапс митрального клапана;
- кальциноз митрального кольца (до 40 лет);
- расширение легочного ствола (до 40 лет);
- расширение или расслаивающая аневризма грудной или брюшной аорты (до 50 лет).
- О вовлечении сердечно-сосудистой системы говорят, если имеется любой из малых критериев.
- Дыхательная система.
- Больших критериев нет.
- Малые критерии: спонтанный пневмоторакс; буллы в верхушке легкого.
- О вовлечении дыхательной системы говорят, если имеется хотя бы один малый критерий.
- Нервная система.
- Большой критерий:
- Эктазия твердой мозговой оболочки в пояснично-крестцовом отделе (выявляется с помощью КТ или МРТ).
- Малых критериев нет.
- Кожа.
- Больших критериев нет.
- Малые критерии:
- стрии, несвязанные с ожирением;
- рецидивирующие или послеоперационные грыжи.
- О вовлечении кожи говорят, если имеется хотя бы один малый критерий.
- Генетические критерии.
- Большие критерии:
- клинически диагностированный синдром Марфана у родителя, ребенка, брата или сестры больного;
- мутация гена FBN1 у больного;
- наличие у больного тех же генетических маркеров вокруг гена FBN1, что и у родственника с подтвержденным синдромом Марфана.
Малых критериев нет.
Течение и прогноз. Детям и подросткам необходима профилактика сколиоза. Сердечно-сосудистые осложнения могут возникать на любом этапе жизни больного, от внутриутробного до старческого, и выступают основной причиной смерти.
Первым обычно поражается митральный клапан, так что еще до сильного расширения аорты может потребоваться операция по поводу митральной недостаточности.
Дети с небольшим расширением восходящей аорты не нуждаются в ограничении физической активности, хотя тяжелых нагрузок, в том числе участия в спортивных соревнованиях, все же следует избегать. Возможно развитие вторичной глаукомы, особенно при смещении хрусталика в переднюю камеру глаза.
Наиболее высок риск расслаивания аорты у женщин в третьем триместре беременности, в период родов и первый месяц после родов. Сейчас, благодаря ранней профилактике осложнений, продолжительность жизни больных приближается к общепопуляционной.
Синдром Марфана наследуется по аутосомно-доминантному типу и проявляется весьма разнообразно, так что ее бывает сложно заподозрить у больных с неотягощенным семейным анамнезом. Заболевание характеризуется различной пенетрантностью и экспрессивностью. Примерно в 75% случаев носит семейный характер и лишь 25% случаев вызываются спорадическими мутациями.
Причиной синдрома Марфана являются мутации в гене FBN1, который размещается на длинном плече 15 хромосомы в локусе 15q 21.1.
Продукт этого гена, белок фибриллин-1, представляет собой гликопротеид, имеет важное значение для правильного формирования внеклеточного матрикса, играет определенную роль при биогенезе и влияет на функционирование эластических волокон.
Кроме того, внеклеточный матрикс обеспечивает структурную целостность соединительной ткани и играет роль резервуара для факторов роста (класс небольших природных пептидов и белков), главной целью которых является стимулирование роста клеток.
Эластических волокон очень много во всем организме человека, но сконцентрированы они в основном в аорте, связках, в частности в цинновой связке (особая связка, с помощью которой хрусталик прикрепляется к цилиарному телу), именно поэтому эти части организма повреждаются при болезни Марфана в первую очередь. При нарушении структуры микрофибрилл эластических волокон соединительная ткань приобретает повышенную способностью к растяжению и становится менее вынослива к физическим нагрузкам.
Ген FBN1 состоит из 65 кодирующих и одного некодирующего экзона.
Последовательность белка профибриллина, экспрессирующегося с данного гена, представлена 2871 аминокислотным остатком, которые (за исключением сигнального пептида) образуют 5 структурно различных регионов.
Основной патогенетический механизм проявления мутаций – доминант-негативный эффект: продукты экспрессии с патологических аллелей участвуют в формировании микрофибрилл и нарушают их структуру.
Для мутаций в гене FBN1 выявлены некоторые генотип-фенотип корреляции: показано, что около 40% мутаций в 7 терминальных экзонах (59-65) ассоциированы с мягким фенотипом, в то время как мутации в экзонах 24-32 ассоциированы с классическим, нетипично тяжелым и неонатальным синдромом Марфана.
В 2004 году Т. Mizuguchi (и др.) сообщили, что причиной СМ может быть нарушение обмена трансформирующего фактора роста B (ТGFRB2). Бельгийские ученые Барт Лоец и др.
в 2006 году сообщили, что мутации в TGFBR1 являются причиной аневризмы аорты и марфаноидного фенотипа (Loeys-Dietz синдром).
TGFB имеет негативное влияние на сосудистый тонус гладких мышц и нарушает развитие целостного внеклеточного матрикса: накопление избыточного количества TGFB в легких, клапанах сердца и в аорте ослабляет ткани и вызывает симптомы болезни Марфана.
Сегодня описано около 1254 мутаций в гене FBN1 в разных семьях. При этом большая часть мутаций – это миссенс-мутации (на их долю приходится около 57%), 21% — мелкие инсерции/делеции, около 12% — мутации сайтов сплайсинга, 8% — нонсенс мутации, около 2% мутаций представлены крупными делециями/инсерциями, другими перестройками.
В Центре Молекулярной Генетики проводится прямая ДНК-диагностика синдрома Марфана, основанная на поиске мутаций в «горячих» участках (экзоны 24-32) и во всей кодирующей области и прилежащих интронных областях гена FBN1, методом прямого автоматического секвенирования.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 4.54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Синдром Марфана в практике терапевта и семейного врача: диагностика, тактика ведения, лечение, беременность и роды
К.м.н. И.А. Викторова, профессор Г.И. Нечаева
Омская государственная медицинская академия
Среди всех наследственных заболеваний соединительной ткани наибольший интерес
для терапевтов и врачей общей практики представляет синдром Марфана, так как
продолжительность жизни этих больных ограничена 30–40 годами [1] и у одного
пациента может быть столько проблем со здоровьем, сколько специалистов в
поликлинике. Поскольку заболевание имеет заведомо серьезный прогноз для жизни и
трудоспособности пациентов, установление диагноза накладывает особую
ответственность на врача при первой встрече с больным.
В 1896 году французский профессор–педиатр Антонио Марфан впервые представил
клиническое наблюдение 5–летней девочки Габриель с необычными, непрерывно
прогрессирующими аномалиями скелета [2]. Девочка умерла в юном возрасте,
вероятно, от туберкулеза [3]. Внешний габитус Габриель и подобных пациентов с
тех пор стали именовать синдромом Марфана.
Как позднее выяснилось, в
действительности Габриель страдала врожденной контрактурной арахнодактилией [4].
Через 20 лет были описаны первые фенокопии марфаноподобных синдромов, в
частности, синдрома эктопии хрусталиков с аутосомно–доминантным наследованием
[5,6], еще через 30 лет – синдрома дилатации [7] и расслоения аорты [8],
пролапса митрального клапана [9,10], эктазии твердой мозговой оболочки [11].
Weve H.
[6] впервые предположил, что причиной синдрома Марфана является
дефект мезодермы, а известный американский генетик McKusick в расписании
наследственных болезней человека «On–Line Mendelian Inheritance In Man» (OMIM)
открыл этим синдромом новую нозологическую страницу наследственных заболеваний
соединительной ткани [12]. Фенотип синдрома характеризуется некоторой
протяженностью, начиная от легких, «мягких» проявлений соединительнотканой
дисплазии, встречающихся также и в общей популяции, до случаев с угрожающими
жизни системными расстройствами [13].
Основной документ, на котором базировался диагноз синдрома Марфана, был
представлен в 1986 году – это так называемая Berlin Nosology [14]. Среди
критериев Берлинской Нозологии превыше всего ставились достижения молекулярной
генетики [15].
Однако установленная локализация гена синдрома Марфана в аутосоме
15q21 [16,17], кодирующего микрофибриллярный белок фибриллин–1, не является
единственной и характерной исключительно для синдрома Марфана [18].
Мутация в
гене родственного протеина – фибриллина–2 также ведет к клиническим проявлениям
марфаноидного габитуса [14,15]. Нозологические формы с фенотипом «Марфана»,
такие как контрактурная арахнодактилия и семейный пролапс митрального клапана –
MASS–фенотип, имели мутации в тех же генах [19].
Большинство ошибочных диагнозов
у родственников больных, как оказалось, связаны с переоценкой значимости
молекулярно–генетических исследований, так как в случае их позитивности в
семейной истории болезни приводили к предвзятости диагноза у других членов семьи
[20,21].
Только совместные молекулярно–генетические [22,23,24] и
клинические исследования [25,26] имеют достаточные основания для создания
полноценных диагностических критериев.
Современные критерии диагноза синдрома Марфана разработаны в 1996 году
совместными усилиями генетиков и клиницистов и предлагаются к широкому
использованию врачами всех специальностей [27].
«Большим» критерий считается вследствие его большей специфичности, так как он
редко встречается при других состояниях и в общей популяции.
В целом
диагностическое решение должно приниматься на основании больших критериев
болезни.
Важно отличать «большой критерий», имеющийся в системе органов и
определяющий данное заболевание, от «системы органов, вовлеченной в процесс
соединительнотканой дисплазии».
Диагностические критерии патологии скелета
Большие критерии
. «Большим критерием» патологии скелета считается
наличие не менее 4 из следующих признаков:
- – килевидная деформация грудной клетки или воронкообразная деформация грудной
клетки больших степеней, подлежащая оперативному лечению; - – уменьшение верхнего сегмента тела (рост сидя) по отношению к нижнему или
если размах рук превышает рост на 5%; - – положительные тесты запястья и большого пальца (см. ниже);
- – сколиоз более 20° или спондилолистез;
- – невозможность полного разгибания локтевых суставов (угол < 170°);
- – медиальное смещение внутренних лодыжек в результате продольного
плоскостопия; - – протрузия вертлужной впадины любой степени (при рентгенографии).
Малые критерии:
- – воронкообразная деформация грудной клетки умеренной степени;
- – гипермобильность суставов;
- – высокое аркообразное нёбо со «скученностью» зубов;
- – аномалии черепа и лица (долихоцефалия, гипоплазия скул, эндофтальмия –
глубоко посаженные глаза, ретрогнатия, косо опущенные складки век). - Патология скелета для верификации диагноза «синдром Марфана» должна быть
представлена двумя большими критериями (при наличии всех признаков) или одним
большим критерием (4 признака) и двумя малыми критериями [27].
Cиндром Марфана в практике терапевта и семейного врача: диагностика, тактика ведения, лечение, беременность и роды
Омская государственная медицинская академия
Омская государственная медицинская академия
Среди всех наследственных заболеваний соединительной ткани наибольший интерес для терапевтов и врачей общей практики представляет синдром Марфана, так как продолжительность жизни этих больных ограничена 30–40 годами [1] и у одного пациента может быть столько проблем со здоровьем, сколько специалистов в поликлинике. Поскольку заболевание имеет заведомо серьезный прогноз для жизни и трудоспособности пациентов, установление диагноза накладывает особую ответственность на врача при первой встрече с больным.
В 1896 году французский профессор–педиатр Антонио Марфан впервые представил клиническое наблюдение 5–летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета [2]. Девочка умерла в юном возрасте, вероятно, от туберкулеза [3]. Внешний габитус Габриель и подобных пациентов с тех пор стали именовать синдромом Марфана.
Как позднее выяснилось, в действительности Габриель страдала врожденной контрактурной арахнодактилией [4].
Через 20 лет были описаны первые фенокопии марфаноподобных синдромов, в частности, синдрома эктопии хрусталиков с аутосомно–доминантным наследованием [5,6], еще через 30 лет – синдрома дилатации [7] и расслоения аорты [8], пролапса митрального клапана [9,10], эктазии твердой мозговой оболочки [11].
Weve H.
[6] впервые предположил, что причиной синдрома Марфана является дефект мезодермы, а известный американский генетик McKusick в расписании наследственных болезней человека «On–Line Mendelian Inheritance In Man» (OMIM) открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани [12]. Фенотип синдрома характеризуется некоторой протяженностью, начиная от легких, «мягких» проявлений соединительнотканой дисплазии, встречающихся также и в общей популяции, до случаев с угрожающими жизни системными расстройствами [13].
Основной документ, на котором базировался диагноз синдрома Марфана, был представлен в 1986 году – это так называемая Berlin Nosology [14]. Среди критериев Берлинской Нозологии превыше всего ставились достижения молекулярной генетики [15].
Однако установленная локализация гена синдрома Марфана в аутосоме 15q21 [16,17], кодирующего микрофибриллярный белок фибриллин–1, не является единственной и характерной исключительно для синдрома Марфана [18].
Мутация в гене родственного протеина – фибриллина–2 также ведет к клиническим проявлениям марфаноидного габитуса [14,15]. Нозологические формы с фенотипом «Марфана», такие как контрактурная арахнодактилия и семейный пролапс митрального клапана – MASS–фенотип, имели мутации в тех же генах [19].
Большинство ошибочных диагнозов у родственников больных, как оказалось, связаны с переоценкой значимости молекулярно–генетических исследований, так как в случае их позитивности в семейной истории болезни приводили к предвзятости диагноза у других членов семьи [20,21].
Только совместные молекулярно–генетические [22,23,24] и клинические исследования [25,26] имеют достаточные основания для создания полноценных диагностических критериев.
Современные критерии диагноза синдрома Марфана разработаны в 1996 году совместными усилиями генетиков и клиницистов и предлагаются к широкому использованию врачами всех специальностей [27].
«Большим» критерий считается вследствие его большей специфичности, так как он редко встречается при других состояниях и в общей популяции.
В целом диагностическое решение должно приниматься на основании больших критериев болезни.
Важно отличать «большой критерий», имеющийся в системе органов и определяющий данное заболевание, от «системы органов, вовлеченной в процесс соединительнотканой дисплазии».
Диагностические критерии патологии скелета
Большие критерии. «Большим критерием» патологии скелета считается наличие не менее 4 из следующих признаков:
- – килевидная деформация грудной клетки или воронкообразная деформация грудной клетки больших степеней, подлежащая оперативному лечению;
- – уменьшение верхнего сегмента тела (рост сидя) по отношению к нижнему или если размах рук превышает рост на 5%;
- – положительные тесты запястья и большого пальца (см. ниже);
- – сколиоз более 20° или спондилолистез;
- – невозможность полного разгибания локтевых суставов (угол < 170°);
- – медиальное смещение внутренних лодыжек в результате продольного плоскостопия;
- – протрузия вертлужной впадины любой степени (при рентгенографии).
- Малые критерии:
- – воронкообразная деформация грудной клетки умеренной степени;
- – гипермобильность суставов;
- – высокое аркообразное нёбо со «скученностью» зубов;
- – аномалии черепа и лица (долихоцефалия, гипоплазия скул, эндофтальмия – глубоко посаженные глаза, ретрогнатия, косо опущенные складки век).
- Патология скелета для верификации диагноза «синдром Марфана» должна быть представлена двумя большими критериями (при наличии всех признаков) или одним большим критерием (4 признака) и двумя малыми критериями [27].